Nach der Destillation will man ja oft wissen, wie viel Alkohol in der Destille war. Also zum Beispiel, wie viel Alkohol die Maische hatte. Hierbei hilft der neue Rechner:
vol% des Destilleninhalts
Zusätzlich wird auch die Menge und Alkoholstärke der verbleibenden Destillenflüssigkeit (Schlempe, Backset, Dunder) berechnet.
Rechner
-
Hügelwilli

- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Re: Rechner
Wenn man beim Destillieren wissen möchte, welche vol% denn die angezeigten Dampftemperaturen genau bedeuten, kann man sich das zwar mit unserem Destillationsrechner anzeigen lassen, einfacher ist es aber mit einer Tabelle.
Wir haben deswegen nun einen Rechner, welcher pdfs mit Destillationsdaten erstellt:
Destillationstabellen-Generator
Nach Eingabe des Luftdrucks wird eine Tabelle mit der Dampftemperatur in 0.1°C-Schritten und den dazugehörien vol% im Destillat angezeigt. Diese kann auch als pdf abgespeichert und ausgedruckt werden.
Die Tabelle gilt für alle Destillen, auch für Refluxdestillen, wenn das Thermometer über der Packung, also nach der Rektifikation, angebracht ist.
Man beachte bitte die Anmerkung unter dem Rechner über das Herausfinden des Luftdrucks.
Diese Anmerkung und auch die dazugehörige Rechenweise sind neu und hat nun auch in dem Destillationsrechner und dem Destillationsrechner Molverhältnis die alte etwas fehlerhafte Rechenweise und Anmerkung ersetzt.
Wir haben deswegen nun einen Rechner, welcher pdfs mit Destillationsdaten erstellt:
Destillationstabellen-Generator
Nach Eingabe des Luftdrucks wird eine Tabelle mit der Dampftemperatur in 0.1°C-Schritten und den dazugehörien vol% im Destillat angezeigt. Diese kann auch als pdf abgespeichert und ausgedruckt werden.
Die Tabelle gilt für alle Destillen, auch für Refluxdestillen, wenn das Thermometer über der Packung, also nach der Rektifikation, angebracht ist.
Man beachte bitte die Anmerkung unter dem Rechner über das Herausfinden des Luftdrucks.
Diese Anmerkung und auch die dazugehörige Rechenweise sind neu und hat nun auch in dem Destillationsrechner und dem Destillationsrechner Molverhältnis die alte etwas fehlerhafte Rechenweise und Anmerkung ersetzt.
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Re: Rechner, die Rektifikationszahl
Bei beiden Destillationssimulatoren kann man einen Wert für die Rektifikation eintragen. Ich schreibe hier ein bisschen darüber, was diese Angabe bedeutet, denn wir arbeiten gerade an einem neuen Rechner, einem unter anderem die Rektifikation berücksichtigenden Siedediagramm-Generator, welcher dafür eine genau Definition verlangt:
"0" bedeutet keine Rektifikation, also eine einfache Destillation.
"1" bedeutet, daß das Destillat in einem Durchgang zweimal destilliert wird. Und zwar genau zweimal. Nicht im Schnitt zweimal. Im Schnitt zweimal wäre von der Reinheit des Destillats niedriger als alles genau zweimal, da jede weitere Destillation nicht so viel bringt wie die vorige. Anders gesagt: 1 Teil einmal, 2 Teile zweimal und 1 Teil dreimal destilliert, also im Schnitt zweimal, endet bei niedrigeren vol% als alle 4 Teile zweimal destilliert.
Es ist eine reine Definitionsfrage. Wir haben uns für diese Definition vor allem entschieden, weil sie genau berechenbar ist.
"0.3" Rektifikation bedeutet bei uns, daß von 1kg Destillat 0.3kg zweifach und 0.7kg einfach destilliert wurden. Das haben wir vor kurzem nochmal ändern müssen. Also die Simulatoren zeigen jetzt bei Rektifikationszahlen mit Kommastellen ein bisschen andere Ergebnisse an. Aber so wird es jetzt hoffentlich bleiben.
Der einzige bisher bemerkte Nachteil unserer Definitionen:
Der Unterschied zum Beispiel zwischen 0.9 und 1.0 Rektifikation ist wesentlich größer als der Unterschied zwischen 1.0 und 1.1 Rektifikation. Das ist auf den erste Blick extrem unlogisch, erklärt sich aber durch unsere Definition:
0.9 Rektifikation bedeutet 9 Teile zweifach und 1 Teil einfach destilliert.
1.1 Rektifikation bedeutet 9 Teile zweifach und 1 Teil dreifach destilliert.
Der 1 Teil einfach bei 0.9 verursacht nun aber viel mehr Unreinheit, als der 1 Teil dreifach bei 1.1 mehr Reinheit bringt. Daher ist der Abstand zwischen 0.9 und 1.0 wesentlich größer, als der zwischen 1.0 und 1.1. ZB mit 10vol% in der Destille bedeuten:
0.8: 77.3vol% im Destillat
0.9: 79.8vol%
1.0: 82.3vol%
1.1: 83.0vol%
1.2: 83.6vol%
Man sieht, wie der Abstand der vol% von 1.0 auf 1.1 plötzlich kleiner ist.
Wäre unsere Definition "im Schnitt", wäre die Reinigungssteigerung pro 0.1 Rektifikation viel logischer. Das wäre ein Vorteil dieser Definition. Bei unserer Definition dagegen gibt es mehr oder weniger große Brüche nach jeder ganzen Zahl.
Edit: Dieses Problem ist zum größten Teil erledigt, weil wir die Siedediagrammdaten inzwischen erweitert haben. Wir haben nun Datenreihen auch für 0.1, 0.2, 0.3 ... 0.9 Destillationsstufen, die wir so gestaltetn haben, daß diese plötzlichen Sprünge nun weg sind.
Im Endeffekt haben beide Simulatoren immer recht. Wenn nicht, war die Angabe der Rektifikationszahl halt falsch. Das hilft natürlich wenig... Der einzige Weg ist, die eigenen Vermutungen über die Rektifikationszahl immer wieder mit den Rechenergebnissen zu vergleichen und zu korrigieren und dadurch dazuzulernen.
Wenn man denn überhaupt an Berechnungen dieser Art interessiert ist. Guten Schnaps machen kann man durchaus auch ohne übrigens.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
"0" bedeutet keine Rektifikation, also eine einfache Destillation.
"1" bedeutet, daß das Destillat in einem Durchgang zweimal destilliert wird. Und zwar genau zweimal. Nicht im Schnitt zweimal. Im Schnitt zweimal wäre von der Reinheit des Destillats niedriger als alles genau zweimal, da jede weitere Destillation nicht so viel bringt wie die vorige. Anders gesagt: 1 Teil einmal, 2 Teile zweimal und 1 Teil dreimal destilliert, also im Schnitt zweimal, endet bei niedrigeren vol% als alle 4 Teile zweimal destilliert.
Es ist eine reine Definitionsfrage. Wir haben uns für diese Definition vor allem entschieden, weil sie genau berechenbar ist.
"0.3" Rektifikation bedeutet bei uns, daß von 1kg Destillat 0.3kg zweifach und 0.7kg einfach destilliert wurden. Das haben wir vor kurzem nochmal ändern müssen. Also die Simulatoren zeigen jetzt bei Rektifikationszahlen mit Kommastellen ein bisschen andere Ergebnisse an. Aber so wird es jetzt hoffentlich bleiben.
Der einzige bisher bemerkte Nachteil unserer Definitionen:
Der Unterschied zum Beispiel zwischen 0.9 und 1.0 Rektifikation ist wesentlich größer als der Unterschied zwischen 1.0 und 1.1 Rektifikation. Das ist auf den erste Blick extrem unlogisch, erklärt sich aber durch unsere Definition:
0.9 Rektifikation bedeutet 9 Teile zweifach und 1 Teil einfach destilliert.
1.1 Rektifikation bedeutet 9 Teile zweifach und 1 Teil dreifach destilliert.
Der 1 Teil einfach bei 0.9 verursacht nun aber viel mehr Unreinheit, als der 1 Teil dreifach bei 1.1 mehr Reinheit bringt. Daher ist der Abstand zwischen 0.9 und 1.0 wesentlich größer, als der zwischen 1.0 und 1.1. ZB mit 10vol% in der Destille bedeuten:
0.8: 77.3vol% im Destillat
0.9: 79.8vol%
1.0: 82.3vol%
1.1: 83.0vol%
1.2: 83.6vol%
Man sieht, wie der Abstand der vol% von 1.0 auf 1.1 plötzlich kleiner ist.
Wäre unsere Definition "im Schnitt", wäre die Reinigungssteigerung pro 0.1 Rektifikation viel logischer. Das wäre ein Vorteil dieser Definition. Bei unserer Definition dagegen gibt es mehr oder weniger große Brüche nach jeder ganzen Zahl.
Edit: Dieses Problem ist zum größten Teil erledigt, weil wir die Siedediagrammdaten inzwischen erweitert haben. Wir haben nun Datenreihen auch für 0.1, 0.2, 0.3 ... 0.9 Destillationsstufen, die wir so gestaltetn haben, daß diese plötzlichen Sprünge nun weg sind.
Im Endeffekt haben beide Simulatoren immer recht. Wenn nicht, war die Angabe der Rektifikationszahl halt falsch. Das hilft natürlich wenig... Der einzige Weg ist, die eigenen Vermutungen über die Rektifikationszahl immer wieder mit den Rechenergebnissen zu vergleichen und zu korrigieren und dadurch dazuzulernen.
Wenn man denn überhaupt an Berechnungen dieser Art interessiert ist. Guten Schnaps machen kann man durchaus auch ohne übrigens.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Re: Rechner, Siedediagramm-Generator
Ein neues Spielzeug ist da!
Siedediagramm-Generator
Nach Eingabe des Luftdrucks oder der Höhenmeter wird ein Diagramm erstellt. Für die, die nicht wissen, was es bedeutet, ist es hier erklärt.
Man kann auch die Rektifikation eingeben. Dann werden zusätzlich bunte Balken in die Siedelinse gezeichnet, an denen man die durch die Rektifikation erhöhte Destillationsleistung ablesen kann.
Dieses Diagramm ist interaktiv. Das heißt man kann den Cursor auf die Achsenlinien führen, dann werden Linien von dort zu den anderen Werten gezogen. Also zB:
Hier liegt der Cursor auf 95°C. Das ist die Temperatur in der Destille. Die Linien führen nach unten zu der Alkoholstärke in der Destille (6.1vol%), nach rechts oben zu der Alkohlstärke des Destillats (62.3vol%) und rechts zur Dampftemperatur (90°C).
Hier das gleiche mit 0 Rektifikation:
Weil nicht rektifiziert wird, werden nur 42.7vol% erreicht und die Dampftemperatur entspricht der Siedetemperatur.
Man kann das Diagramm auch als pdf herunterladen und ausdrucken. Ohne die interaktiven Linien.
Was kann man von den Diagrammen lernen?
Zum Beispiel, daß es bei hoher Rektifikation so gut wie egal ist, wie viel Alkohol man im Kessel hat. Zumindest bezogen auf die erreichten vol% im Destillat. Hier 700 hPa und Rektifikation 9, also eine zehnfache Destillation. 50vol% im Kessel ergeben etwa 96.6vol% im Destillat:
Nur 0.5vol% im Kessel ergeben etwa 96.1vol% im Destillat:
Die hundertfache Alkoholkonzentration in der Destille bringt also nur 0.5vol% mehr im Destillat.
Das Diagramm erklärt also den Sachverhalt, daß bei einer Refluxdestille die vol% ewig oben bleiben und erst absacken, wenn kein Alkohol mehr da ist.
Auch bei niedriger Rektifikation ist das im Prinzip schon sichtbar: Zum Beispiel bei 0.5 Rektifikation, also einer langsam laufende Potstill mit langem gefüllten Steigrohr:
Wenn man nun mit dem Cursor unten bei 30 oder 40vol% in der Destille anfängt (also wie bei einem Feinbrand) und ihn langsam und gleichmäßig Richtung 0vol% bewegt, erkennt man wie sich die beiden blauen Linien des Destillats (rechts oben) anfangs nur wenig bewegen und dann aber immer schneller.
Das Diagramm stellt also das typische Potstillverhalten beim Feinbrand da. Nämlich, daß die vol% recht lange stabil im oberen Bereich bleiben, dann aber immer schneller absacken.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
Siedediagramm-Generator
Nach Eingabe des Luftdrucks oder der Höhenmeter wird ein Diagramm erstellt. Für die, die nicht wissen, was es bedeutet, ist es hier erklärt.
Man kann auch die Rektifikation eingeben. Dann werden zusätzlich bunte Balken in die Siedelinse gezeichnet, an denen man die durch die Rektifikation erhöhte Destillationsleistung ablesen kann.
Dieses Diagramm ist interaktiv. Das heißt man kann den Cursor auf die Achsenlinien führen, dann werden Linien von dort zu den anderen Werten gezogen. Also zB:
Hier das gleiche mit 0 Rektifikation:
Man kann das Diagramm auch als pdf herunterladen und ausdrucken. Ohne die interaktiven Linien.
Was kann man von den Diagrammen lernen?
Zum Beispiel, daß es bei hoher Rektifikation so gut wie egal ist, wie viel Alkohol man im Kessel hat. Zumindest bezogen auf die erreichten vol% im Destillat. Hier 700 hPa und Rektifikation 9, also eine zehnfache Destillation. 50vol% im Kessel ergeben etwa 96.6vol% im Destillat:
Das Diagramm erklärt also den Sachverhalt, daß bei einer Refluxdestille die vol% ewig oben bleiben und erst absacken, wenn kein Alkohol mehr da ist.
Auch bei niedriger Rektifikation ist das im Prinzip schon sichtbar: Zum Beispiel bei 0.5 Rektifikation, also einer langsam laufende Potstill mit langem gefüllten Steigrohr:
Das Diagramm stellt also das typische Potstillverhalten beim Feinbrand da. Nämlich, daß die vol% recht lange stabil im oberen Bereich bleiben, dann aber immer schneller absacken.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Re: Rechner, McCabe-Thiele-Verfahren
Ein neuer Rechner ist da. Oder besser gesagt: Ein Package aus sechs! neuen Rechnern für Refluxdestillationen.
Bei allen geht es um das McCabe-Thiele-Verfahren.
Mit diesem kann man den Zusammenhang von vier Parametern berechnen. Von:
1. der Alkoholstärke in der Destille,
2. der Alkoholstärke im Destillat,
3. der Anzahl der Böden und
4. dem Rückflussverhältnis.
Sind drei der vier Werte bekannt, lässt sich der vierte ermitteln.
Genau dies macht der McCabe-Thiele-Verfahren-Hauptrechner.
Zusätzlich berechnet er, wie viele theoretische Destillationen dabei stattfinden, bzw die Zahl "Rektifikation", welche bei unseren beiden Destillationssimulatoren angegeben werden muss.
Das Interessante an diesem Verfahren ist, daß es Berechnungen mit dem eingestellten Rückflussverhältnis und des nötigen Rückflussverhältnisses ermöglicht, was wir bisher noch nicht konnten. Das eröffnet uns auch neue Möglichkeiten.
Von dem Hauptrechner kommt man zu vier "praxisbezogenen Rechnervarianten". Diese machen das gleiche wie der Hauptrechner, sind aber für den jeweiligen Zweck (welcher der vier Parameter berechnet werden soll) benutzerfreundlicher gestaltet.
Und man kommt noch zu "McCabe-Thiele-Diagramme", wo man sich leere McCabe-Thiele-Diagramme anschauen und als pdf herunterladen kann. Drei Diagramme: Bezogen auf vol%, gew% und Molanteil. Dieses Verfahren ist nämlich ein graphisches, erklärt sich visualisiert also am besten.
So kann ein ausgefülltes Diagramm ausschauen:
Eine kurze Erklärung dazu:
Die senkrechte blaue Gerade links ist die Alkoholstärke in der Destille (ca. 7gew%), die blaue senkrechte weiter rechts die Alkoholstärke im Destillat (85gew%), die Steigung der grünen "Arbeitsgerade" ist das Rückflussverhältnis (wäre sie waagrecht, würde das 0% Reflux bedeuten, wäre sie 45°, also würde sie auf der schwarzen schrägen Gerade liegen, würde das 100% Reflux bedeuten) und die Anzahl der Treppenstufen die Anzahl der theoretischen Böden (vier).
Genauere Erklärungen finden sich sehr viele im Netz, da dieses Verfahren sehr wichtig für die Industrie ist. Meist geht es aber um kontinuierliche Destillation. Um dieses Verfahren für unsere nicht-kontinuierlichen Destillen erklärt zu bekommen, muss man nach "batch distillation McCabe Thiele method" suchen.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
Bei allen geht es um das McCabe-Thiele-Verfahren.
Mit diesem kann man den Zusammenhang von vier Parametern berechnen. Von:
1. der Alkoholstärke in der Destille,
2. der Alkoholstärke im Destillat,
3. der Anzahl der Böden und
4. dem Rückflussverhältnis.
Sind drei der vier Werte bekannt, lässt sich der vierte ermitteln.
Genau dies macht der McCabe-Thiele-Verfahren-Hauptrechner.
Zusätzlich berechnet er, wie viele theoretische Destillationen dabei stattfinden, bzw die Zahl "Rektifikation", welche bei unseren beiden Destillationssimulatoren angegeben werden muss.
Das Interessante an diesem Verfahren ist, daß es Berechnungen mit dem eingestellten Rückflussverhältnis und des nötigen Rückflussverhältnisses ermöglicht, was wir bisher noch nicht konnten. Das eröffnet uns auch neue Möglichkeiten.
Von dem Hauptrechner kommt man zu vier "praxisbezogenen Rechnervarianten". Diese machen das gleiche wie der Hauptrechner, sind aber für den jeweiligen Zweck (welcher der vier Parameter berechnet werden soll) benutzerfreundlicher gestaltet.
Und man kommt noch zu "McCabe-Thiele-Diagramme", wo man sich leere McCabe-Thiele-Diagramme anschauen und als pdf herunterladen kann. Drei Diagramme: Bezogen auf vol%, gew% und Molanteil. Dieses Verfahren ist nämlich ein graphisches, erklärt sich visualisiert also am besten.
So kann ein ausgefülltes Diagramm ausschauen:
Die senkrechte blaue Gerade links ist die Alkoholstärke in der Destille (ca. 7gew%), die blaue senkrechte weiter rechts die Alkoholstärke im Destillat (85gew%), die Steigung der grünen "Arbeitsgerade" ist das Rückflussverhältnis (wäre sie waagrecht, würde das 0% Reflux bedeuten, wäre sie 45°, also würde sie auf der schwarzen schrägen Gerade liegen, würde das 100% Reflux bedeuten) und die Anzahl der Treppenstufen die Anzahl der theoretischen Böden (vier).
Genauere Erklärungen finden sich sehr viele im Netz, da dieses Verfahren sehr wichtig für die Industrie ist. Meist geht es aber um kontinuierliche Destillation. Um dieses Verfahren für unsere nicht-kontinuierlichen Destillen erklärt zu bekommen, muss man nach "batch distillation McCabe Thiele method" suchen.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Re: Rechner, Kolonnensimulator
Wir haben nun einen Kolonnensimulator!
Im Endeffekt beruht er auf dem MacCabe-Thiele-Rechner. Er ist aber besser bedienbar(Drehregler) und vor allem wird die ganze Kolonne -also alle Böden- berechnet und graphisch dargestellt. Angegeben werden muss die Alkoholstärke im Kessel, die Anzahl der Böden und wie viel Prozent des Dampfes als Produkt abgezogen werden. Zusätzlich der Luftdruck (bitte Anmerkungen unter dem Rechner beachten).
Macht Spaß das Spielzeug. Aber nicht nur das. Phänomene, wie zum Beispiel das urplötzliche Absacken des Alkoholgehalts am Ende der Reflux-Destillation bei sehr tief gesunkenen vol% im Kessel, sind sichtbar. Mit Herumspielen an den Reglern kann man doch einiges lernen.
Zum Beispiel hier ist noch alles in Ordnung:
Obwohl nur noch 1.3vol% im Kessel sind, erreicht man oben noch 96vol% bei immerhin 10% Entnahme des Dampfes.
Nun aber sind nur noch 1vol% im Kessel und oben sackt die Alkoholstärke auf 81.6vol% ab:
Aber schon ein bisschen weniger Produkt entnehmen (8 anstelle 10% der Dampfmenge) und schon steigt die Alkoholstärke oben wieder auf 96vol%:
Der McCabe-Thiele-Rechner ist nun in den praxisfernen Bereich verschoben.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
Im Endeffekt beruht er auf dem MacCabe-Thiele-Rechner. Er ist aber besser bedienbar(Drehregler) und vor allem wird die ganze Kolonne -also alle Böden- berechnet und graphisch dargestellt. Angegeben werden muss die Alkoholstärke im Kessel, die Anzahl der Böden und wie viel Prozent des Dampfes als Produkt abgezogen werden. Zusätzlich der Luftdruck (bitte Anmerkungen unter dem Rechner beachten).
Macht Spaß das Spielzeug. Aber nicht nur das. Phänomene, wie zum Beispiel das urplötzliche Absacken des Alkoholgehalts am Ende der Reflux-Destillation bei sehr tief gesunkenen vol% im Kessel, sind sichtbar. Mit Herumspielen an den Reglern kann man doch einiges lernen.
Zum Beispiel hier ist noch alles in Ordnung:
Nun aber sind nur noch 1vol% im Kessel und oben sackt die Alkoholstärke auf 81.6vol% ab:
Aber schon ein bisschen weniger Produkt entnehmen (8 anstelle 10% der Dampfmenge) und schon steigt die Alkoholstärke oben wieder auf 96vol%:
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Re: Rechner, Thermometerfehler, Luftdruck
Ein neuer Rechner ist da! Er heißt Thermometerfehler
Da ja die meisten hier kein Thermometer haben, welches auf 0.1°C genau anzeigt, haben wir ein kleines Tool gebastelt, wo man den Luftdruck und die angezeigte Dampftemperatur bei einer Destillation von Wasser eingeben kann. Berechnet wird dann, wie viel das Thermometer zu tief oder zu hoch anzeigt.
Diesen Wert kann man nun in manche unserer Rechner eingeben, er wird dann bei der Berechnung berücksichtigt. Sowohl bei der Berechnung von Temperaturen als auch bei der Eingabe dieser. Ausgegeben werden dann also nicht mehr die realen Temperaturen, sondern die auf dem Thermometer angezeigten, sobald ein Thermometerfehler angegeben wird.
Und wir haben ein paar Rechner überarbeitet:
Einige Rechner erlauben ja die Berücksichtigung des Luftdrucks. Dafür kann man entweder direkt den absoluten Lufdruck eingeben oder die Höhenmeter, Außentemperatur und den relativen Luftdruck.
Die Berücksichtigung des Drucks ist jetzt in weiteren Rechnern eingebaut.
Damit es nicht unübersichtlich wird, ist aber nur noch die Eingabe des absoluten Luftdrucks möglich. Die Berechnung des absoluten Luftdrucks aus den Höhenmetern, dem rel. Luftdruck und der Außentemperatur ist meist nicht nötig, da die meisten ein Smartphone haben, mit dem man sich den absoluten Luftdruck anzeigen lassen kann. Falls jemand diese Berechnung trotzdem braucht, haben wir sie in einen eigenen Rechner verschoben: Luftdruck
Er verwendet eine andere Formel als die Rechner bisher. Die bisherige Formel war zwar nicht schlechter, aber die neue ist die, welche die Wetterdienste verwenden. Das bedeutet, bei Angabe der Höhenmeter mit zusätzlicher Angabe eines relativen Luftdrucks (bezogen von Wetterdiensten) bekommt man rechnerisch genau den Messwert zurück, also den gemessenen absoluten Luftdruck. Anders gesagt, Unzulänglichkeiten der Formel (damit ist vor allem gemeint, daß diese Formeln nicht die ganze Wirklichkeit abbilden, also immer etwas ungenau sein können) heben sich auf, da sie vom Wetterdienst vorwärts und von uns dann rückwärts eingesetzt wurde. Ungenauigkeiten können also nur noch vom Druckmessgerät kommen oder von ungenauen Höhen- oder Temperaturangaben.
Im Endeffekt berechnet die neue Formel eine etwas kleinere Beeinflussung des Luftdrucks durch die Höhenmeter. Macht aber in normalen Höhenlagen kaum was aus.
Was alles geändert wurde:
Destillationsrechner, Destillationsrechner Molverhältnis und Destillationssimulator 2: Berücksichtigung des Drucks und des Thermometerfehlers.
Destillationssimulator 1: Berechnung der Temperaturspannen der Abschnitte und die Möglichkeit der Angabe von Temperaturen als Schnittpunkte. Berücksichtigung des Drucks und des Thermometerfehlers. Außerdem ist er nun etwas besser abgesichert, wenn unmögliche Vorgaben eingegeben werden.
Kolonnensimulator, Destillationstabellen-Generator und Siedediagramm-Generator: Eingabe des Thermometerfehlers.
Da ja die meisten hier kein Thermometer haben, welches auf 0.1°C genau anzeigt, haben wir ein kleines Tool gebastelt, wo man den Luftdruck und die angezeigte Dampftemperatur bei einer Destillation von Wasser eingeben kann. Berechnet wird dann, wie viel das Thermometer zu tief oder zu hoch anzeigt.
Diesen Wert kann man nun in manche unserer Rechner eingeben, er wird dann bei der Berechnung berücksichtigt. Sowohl bei der Berechnung von Temperaturen als auch bei der Eingabe dieser. Ausgegeben werden dann also nicht mehr die realen Temperaturen, sondern die auf dem Thermometer angezeigten, sobald ein Thermometerfehler angegeben wird.
Und wir haben ein paar Rechner überarbeitet:
Einige Rechner erlauben ja die Berücksichtigung des Luftdrucks. Dafür kann man entweder direkt den absoluten Lufdruck eingeben oder die Höhenmeter, Außentemperatur und den relativen Luftdruck.
Die Berücksichtigung des Drucks ist jetzt in weiteren Rechnern eingebaut.
Damit es nicht unübersichtlich wird, ist aber nur noch die Eingabe des absoluten Luftdrucks möglich. Die Berechnung des absoluten Luftdrucks aus den Höhenmetern, dem rel. Luftdruck und der Außentemperatur ist meist nicht nötig, da die meisten ein Smartphone haben, mit dem man sich den absoluten Luftdruck anzeigen lassen kann. Falls jemand diese Berechnung trotzdem braucht, haben wir sie in einen eigenen Rechner verschoben: Luftdruck
Er verwendet eine andere Formel als die Rechner bisher. Die bisherige Formel war zwar nicht schlechter, aber die neue ist die, welche die Wetterdienste verwenden. Das bedeutet, bei Angabe der Höhenmeter mit zusätzlicher Angabe eines relativen Luftdrucks (bezogen von Wetterdiensten) bekommt man rechnerisch genau den Messwert zurück, also den gemessenen absoluten Luftdruck. Anders gesagt, Unzulänglichkeiten der Formel (damit ist vor allem gemeint, daß diese Formeln nicht die ganze Wirklichkeit abbilden, also immer etwas ungenau sein können) heben sich auf, da sie vom Wetterdienst vorwärts und von uns dann rückwärts eingesetzt wurde. Ungenauigkeiten können also nur noch vom Druckmessgerät kommen oder von ungenauen Höhen- oder Temperaturangaben.
Im Endeffekt berechnet die neue Formel eine etwas kleinere Beeinflussung des Luftdrucks durch die Höhenmeter. Macht aber in normalen Höhenlagen kaum was aus.
Was alles geändert wurde:
Destillationsrechner, Destillationsrechner Molverhältnis und Destillationssimulator 2: Berücksichtigung des Drucks und des Thermometerfehlers.
Destillationssimulator 1: Berechnung der Temperaturspannen der Abschnitte und die Möglichkeit der Angabe von Temperaturen als Schnittpunkte. Berücksichtigung des Drucks und des Thermometerfehlers. Außerdem ist er nun etwas besser abgesichert, wenn unmögliche Vorgaben eingegeben werden.
Kolonnensimulator, Destillationstabellen-Generator und Siedediagramm-Generator: Eingabe des Thermometerfehlers.
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Re: Rechner, Thermometerfehler
Den Rechner Thermometerfehler ist überarbeitet und erweitert.
Bisher hat er aus den eingegebenen Daten berechnet, welchen Fehler das Thermometer bei Wasserdampf anzeigt. Die "thermometerfähigen" Rechner, also die, wo man einen Thermometerfehler eingeben kann, gehen dann davon aus, daß dieser Fehler für alle Temperaturen gilt; daß also ein Thermometer, welches bei Wasserdampf 101°C anstelle 100°C anzeigt, dann 1°C bei 0°C anzeigt.
Das ist aber natürlich nicht unbedingt so. Deshalb kann man im neuen Thermometerfehlerrechner nun mit einer Eiswassermessung den Fehler auch für 0°C berechnen. Andere Temperaturen können dann von den entsprechenden Rechnern linear interpoliert werden. Also ein Thermometer, welches bei 0°C 1°C zu viel anzeigt und bei 100°C 1°C zu wenig, zeigt bei 50°C genau richtig an. Das ist mit großer Wahrscheinlichkeit sehr exakt. Der Rechner ersetzt also eine Zweipunktkalibrierung, welche bei unseren Thermometern normalerweise leider nicht möglich ist.
Der neue Thermometerfehler besteht daher nun aus zwei Zahlen, dem Fehler bei 0°C und dem bei 100°C, durch ein Leerzeichen getrennt. Zum Beispiel bedeutet "0.5 -1", daß das Thermometer 0.5°C bei 0°C und 99°C bei 100°C anzeigt.
Wenn man nur eine Zahl eingibt, funktionieren die Rechner aber so wie bisher. Also dieser eine Fehler gilt dann für alle Temperaturen, egal ob er mit Eiswasser oder Wasserdampf ermittelt wurde.
Zusätzlich ist im Thermometerfehlerrechner weiter unten ein kleines Tool, mit dem man anhand des Thermometerfehlers eingegebene Temperaturen korrigieren oder für gewünschte Temperaturen den erforderten Anzeigewert des Thermometers berechnen kann. Dies ist sinnvoll zum Beispiel bei der Benutzung eines Alkoholmeters.
Zur Eiswassermessung gibt es viele Anleitungen im Internet.
Wenn man es auf 0.1°C genau haben möchte, sollte man einige Dinge wirklich ernst nehmen:
- das Eis muss zerkleinert sein.
- das in den Anleitungen beschriebene Verhältnis von Eis zu Wasser halbwegs beachten.
- als Behälter braucht man eine Thermoskanne.
- destilliertes oder demineralisiertes Wasser bzw. Eis nehmen. Weiches Leitungswasser ist vielleicht auch in Ordnung.
- das Eiswasser muss erstmal in der verschlossenen Thermoskanne 30min vorbereitet werden, damit das Wasser 0°C erreicht. Währenddessen immer mal schütteln.
- das Thermometer braucht dann ein paar Minuten im Eiswasser, bis sich die Temperatur nicht mehr ändert.
Zusätzlich könnte man überlegen:
- Rühren oder nicht rühren beim Messen? Da sind wir uns nicht sicher.
- der Sensor soll kein Eis berühren, sondern nur Wasser.
- kein "unterkühltes" Eis nehmen. Also zum Beispiel das Eis vorher bei nur -2°C lagern, wenn das möglich ist. Oder erst gerade frisch eingefrorene Eiswürfel nehmen, welche innen noch flüssig sind. Und zum Ausgleich das Wasser vorkühlen oder weniger Wasser nehmen.
Zur Wasserdampfdestillation bitte diese Hinweise beachten: Thermometer / Position/ Eintauchtiefe
Bisher hat er aus den eingegebenen Daten berechnet, welchen Fehler das Thermometer bei Wasserdampf anzeigt. Die "thermometerfähigen" Rechner, also die, wo man einen Thermometerfehler eingeben kann, gehen dann davon aus, daß dieser Fehler für alle Temperaturen gilt; daß also ein Thermometer, welches bei Wasserdampf 101°C anstelle 100°C anzeigt, dann 1°C bei 0°C anzeigt.
Das ist aber natürlich nicht unbedingt so. Deshalb kann man im neuen Thermometerfehlerrechner nun mit einer Eiswassermessung den Fehler auch für 0°C berechnen. Andere Temperaturen können dann von den entsprechenden Rechnern linear interpoliert werden. Also ein Thermometer, welches bei 0°C 1°C zu viel anzeigt und bei 100°C 1°C zu wenig, zeigt bei 50°C genau richtig an. Das ist mit großer Wahrscheinlichkeit sehr exakt. Der Rechner ersetzt also eine Zweipunktkalibrierung, welche bei unseren Thermometern normalerweise leider nicht möglich ist.
Der neue Thermometerfehler besteht daher nun aus zwei Zahlen, dem Fehler bei 0°C und dem bei 100°C, durch ein Leerzeichen getrennt. Zum Beispiel bedeutet "0.5 -1", daß das Thermometer 0.5°C bei 0°C und 99°C bei 100°C anzeigt.
Wenn man nur eine Zahl eingibt, funktionieren die Rechner aber so wie bisher. Also dieser eine Fehler gilt dann für alle Temperaturen, egal ob er mit Eiswasser oder Wasserdampf ermittelt wurde.
Zusätzlich ist im Thermometerfehlerrechner weiter unten ein kleines Tool, mit dem man anhand des Thermometerfehlers eingegebene Temperaturen korrigieren oder für gewünschte Temperaturen den erforderten Anzeigewert des Thermometers berechnen kann. Dies ist sinnvoll zum Beispiel bei der Benutzung eines Alkoholmeters.
Zur Eiswassermessung gibt es viele Anleitungen im Internet.
Wenn man es auf 0.1°C genau haben möchte, sollte man einige Dinge wirklich ernst nehmen:
- das Eis muss zerkleinert sein.
- das in den Anleitungen beschriebene Verhältnis von Eis zu Wasser halbwegs beachten.
- als Behälter braucht man eine Thermoskanne.
- destilliertes oder demineralisiertes Wasser bzw. Eis nehmen. Weiches Leitungswasser ist vielleicht auch in Ordnung.
- das Eiswasser muss erstmal in der verschlossenen Thermoskanne 30min vorbereitet werden, damit das Wasser 0°C erreicht. Währenddessen immer mal schütteln.
- das Thermometer braucht dann ein paar Minuten im Eiswasser, bis sich die Temperatur nicht mehr ändert.
Zusätzlich könnte man überlegen:
- Rühren oder nicht rühren beim Messen? Da sind wir uns nicht sicher.
- der Sensor soll kein Eis berühren, sondern nur Wasser.
- kein "unterkühltes" Eis nehmen. Also zum Beispiel das Eis vorher bei nur -2°C lagern, wenn das möglich ist. Oder erst gerade frisch eingefrorene Eiswürfel nehmen, welche innen noch flüssig sind. Und zum Ausgleich das Wasser vorkühlen oder weniger Wasser nehmen.
Zur Wasserdampfdestillation bitte diese Hinweise beachten: Thermometer / Position/ Eintauchtiefe
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Re: Rechner, Refraktometer Temperaturkorrektur
Drei neue Rechner sind da!
Refraktometer Temperaturkorrektur Zuckerlösungen
Refraktometer Temperaturkorrektur Alkohollösungen
Refraktometer Justierung
Es geht also diesmal um Refraktometer. Und zwar um die Temperaturabhängigkeit des Brechungsindex:
Wie auch bei einer Spindel ist der Messwert mit einem Refraktometer leider temperaturabhängig. Je höher die Temperatur, desto niedriger ist der Brechungsindex. Und damit ist auch die angezeigte Zucker- oder Akoholkonzentration zu niedrig. Der falsche Anzeigewert muss bei hohen Temperaturen also nach oben korrigiert werden.
Wahrscheinlich liegt das ganz einfach an der Änderung der Dichte. Jedenfalls ist diese Problematik bei Messungen mit dem Refraktometer ähnlich groß wie mit Spindeln oder anderen Dichtemessgeräten.
Und wie auch bei Spindeln ist dieses Verhalten bei Alkohollösungen viel stärker als bei Zuckerlösungen. Und zwar umso mehr, je höher die Alkoholkonzentration ist. Das bedeutet, daß Messungen von hohen Alkoholstärken, welche mit einem Refraktometer ja sowieso schon nicht einfach sind, da die vol%-Skalenstriche im hohen Bereich sehr nah beinander liegen, zusätzlich durch die hohe Temperaturabhängigkeit erschwert werden.
Bei Messungen des Alkoholgehalts muss daher sehr auf die Genauigkeit des Thermometers geachtet werden.
Aber selbst bei sehr genauen Instrumenten stellt sich die Frage, ob das Refraktometer für Alkohollösungen im Bereich 40-65vol% wirklich das geeignete Messinstrument ist, oder ob nicht doch besser eine (gute) Spindel verwendet werden sollte.
Die Rechner können bei Refraktometern mit eingebauter automatischer Temperaturkompensation (ATC) nicht verwendet werden. Diese korrigieren selber durch einen Bimetallstreifen, welcher sich je nach Temperatur krümmt und dadurch die Skala verschiebt. Die Korrektur ist prinzipiell linear (also pro °C recht ähnlich) und entspricht daher nicht den physikalischen Gegebenheiten, vor allem bei Alkoholmessungen, und sehr ungenau. Der korrigierbare Temperaturbereich umfasst auch nur wenige Grad. Wer also im Weinberg schnell mal messen möchte, kann eines mit ATC kaufen. Wer aber zu Hause möglichst genaue Messungen machen möchte, sollte eines ohne ATC nehmen und ein genaues Thermometer und den Rechner verwenden.
Wobei sich der Wunsch nach höherer Genauigkeit ab einem Punkt leider nur noch erfüllen lässt, indem man tiefer in die Tasche greift und zB ein professionelles Abbe-Refraktometer kauft. Oder sehr große Spindeln oder ein Pyknometer mit Feinwaage... (aus dem Nähkästchen: Pyknometer sind ein aktuelles Projekt von uns. Sie sind wahrscheinlich ein guter und günstiger Mittelweg zwischen Billigspindeln und den ganz teuren Spindelsets über 1000€.)
Die zwei Tropfen, die man in ein Refraktometer gibt, sind sehr schnell an die Temperatur des Refraktometers angeglichen. In die Rechner sollte also die Temperatur des Refraktometers, also der Raumluft, nicht die Temperatur der Flüssigkeit eingegeben werden. Und die Raumtemperatur sollte halbwegs konstant sein und das Refraktometer sich schon länger in dem Raum befinden.
Der Rechner für die Justierung richtet sich eher an professionelle Refraktometer, da zumindest bei Temperaturen über 20°C eine Skala mit Brechungsindex unter 1.333 benötigt wird. Bzw. Skalen unter 0 Brix, 1.000 SG oder 0 vol%.
Ich besitze seit kurzem ein altes selber renoviertes Abbe-Refraktometer von Zeiss. Es hat etwa 50€ gekostet und mit viel Glück musste ich keine Ersatzteile oder Spezialwerkzeug besorgen. Sonst wären die Gesamtkosten wohl auf bis zu 100€ gestiegen. Arbeit musste allerdings schon reingesteckt werden. Ich denke, die Genauigkeit ist zwei- bis dreifach im Vergleich zu einem Handgerät. Also schon deutlich besser, aber es ist auch kein Wunderinstrument.
Update:
Der Rechner Rechner Refraktometer Temperaturkorrektur Alkohollösungen hat eine neue Funktion:
Man kann nun auswählen, daß man ein verdünntes Destillat misst. Der Rechner rechnet dann nach der Temperaturkorrektur zurück auf die vol% des unverdünnten Destillats.
Der Sinn dahinter ist, daß man auf diese Weise eine wesentlich genauere Messung von hochprozentigen Destillaten bekommt. Denn bei über 45vol% ist eine Messung mit einem Refraktometer nicht mehr genau. Man sollte also vor der Messung auf 40vol% oder gerne auch wesentlich weniger verdünnen. Wie viel genauer das dann wird als das unverdünnte Destillat zu messen, hängt aber natürlich von der Genauigkeit der Waage ab.
Wenn man im Rechner "Messung von verdünntem Destillat" auswählt, werden drei zusätzliche Eingabefelder zum Ausfüllen erzeugt:
Refraktometer Temperaturkorrektur Zuckerlösungen
Refraktometer Temperaturkorrektur Alkohollösungen
Refraktometer Justierung
Es geht also diesmal um Refraktometer. Und zwar um die Temperaturabhängigkeit des Brechungsindex:
Wie auch bei einer Spindel ist der Messwert mit einem Refraktometer leider temperaturabhängig. Je höher die Temperatur, desto niedriger ist der Brechungsindex. Und damit ist auch die angezeigte Zucker- oder Akoholkonzentration zu niedrig. Der falsche Anzeigewert muss bei hohen Temperaturen also nach oben korrigiert werden.
Wahrscheinlich liegt das ganz einfach an der Änderung der Dichte. Jedenfalls ist diese Problematik bei Messungen mit dem Refraktometer ähnlich groß wie mit Spindeln oder anderen Dichtemessgeräten.
Und wie auch bei Spindeln ist dieses Verhalten bei Alkohollösungen viel stärker als bei Zuckerlösungen. Und zwar umso mehr, je höher die Alkoholkonzentration ist. Das bedeutet, daß Messungen von hohen Alkoholstärken, welche mit einem Refraktometer ja sowieso schon nicht einfach sind, da die vol%-Skalenstriche im hohen Bereich sehr nah beinander liegen, zusätzlich durch die hohe Temperaturabhängigkeit erschwert werden.
Bei Messungen des Alkoholgehalts muss daher sehr auf die Genauigkeit des Thermometers geachtet werden.
Aber selbst bei sehr genauen Instrumenten stellt sich die Frage, ob das Refraktometer für Alkohollösungen im Bereich 40-65vol% wirklich das geeignete Messinstrument ist, oder ob nicht doch besser eine (gute) Spindel verwendet werden sollte.
Die Rechner können bei Refraktometern mit eingebauter automatischer Temperaturkompensation (ATC) nicht verwendet werden. Diese korrigieren selber durch einen Bimetallstreifen, welcher sich je nach Temperatur krümmt und dadurch die Skala verschiebt. Die Korrektur ist prinzipiell linear (also pro °C recht ähnlich) und entspricht daher nicht den physikalischen Gegebenheiten, vor allem bei Alkoholmessungen, und sehr ungenau. Der korrigierbare Temperaturbereich umfasst auch nur wenige Grad. Wer also im Weinberg schnell mal messen möchte, kann eines mit ATC kaufen. Wer aber zu Hause möglichst genaue Messungen machen möchte, sollte eines ohne ATC nehmen und ein genaues Thermometer und den Rechner verwenden.
Wobei sich der Wunsch nach höherer Genauigkeit ab einem Punkt leider nur noch erfüllen lässt, indem man tiefer in die Tasche greift und zB ein professionelles Abbe-Refraktometer kauft. Oder sehr große Spindeln oder ein Pyknometer mit Feinwaage... (aus dem Nähkästchen: Pyknometer sind ein aktuelles Projekt von uns. Sie sind wahrscheinlich ein guter und günstiger Mittelweg zwischen Billigspindeln und den ganz teuren Spindelsets über 1000€.)
Die zwei Tropfen, die man in ein Refraktometer gibt, sind sehr schnell an die Temperatur des Refraktometers angeglichen. In die Rechner sollte also die Temperatur des Refraktometers, also der Raumluft, nicht die Temperatur der Flüssigkeit eingegeben werden. Und die Raumtemperatur sollte halbwegs konstant sein und das Refraktometer sich schon länger in dem Raum befinden.
Der Rechner für die Justierung richtet sich eher an professionelle Refraktometer, da zumindest bei Temperaturen über 20°C eine Skala mit Brechungsindex unter 1.333 benötigt wird. Bzw. Skalen unter 0 Brix, 1.000 SG oder 0 vol%.
Ich besitze seit kurzem ein altes selber renoviertes Abbe-Refraktometer von Zeiss. Es hat etwa 50€ gekostet und mit viel Glück musste ich keine Ersatzteile oder Spezialwerkzeug besorgen. Sonst wären die Gesamtkosten wohl auf bis zu 100€ gestiegen. Arbeit musste allerdings schon reingesteckt werden. Ich denke, die Genauigkeit ist zwei- bis dreifach im Vergleich zu einem Handgerät. Also schon deutlich besser, aber es ist auch kein Wunderinstrument.
Update:
Der Rechner Rechner Refraktometer Temperaturkorrektur Alkohollösungen hat eine neue Funktion:
Man kann nun auswählen, daß man ein verdünntes Destillat misst. Der Rechner rechnet dann nach der Temperaturkorrektur zurück auf die vol% des unverdünnten Destillats.
Der Sinn dahinter ist, daß man auf diese Weise eine wesentlich genauere Messung von hochprozentigen Destillaten bekommt. Denn bei über 45vol% ist eine Messung mit einem Refraktometer nicht mehr genau. Man sollte also vor der Messung auf 40vol% oder gerne auch wesentlich weniger verdünnen. Wie viel genauer das dann wird als das unverdünnte Destillat zu messen, hängt aber natürlich von der Genauigkeit der Waage ab.
Wenn man im Rechner "Messung von verdünntem Destillat" auswählt, werden drei zusätzliche Eingabefelder zum Ausfüllen erzeugt:
- gramm leeres Glas (bei einer Waage mit Tara-Funktion kann hier auch 0 eingegeben werden)
- gramm mit Destillat gefülltes Glas
- gramm mit Destillat und Wasser gefülltes Glas
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Re: Unsere Siedediagrammdaten
Wir hatten vor einiger Zeit entdeckt, daß uns bei der Umrechnung unserer Siedediagrammdaten von mol% in vol% ein kleinen Fehler passiert war.
Das hatten wir zum Anlass genommen, uns die Daten und vor allem die Kurven, welche wir daraus gemacht hatten, also wie wir die Messdaten zu einem vernünftigen Verlauf geglättet hatten, nochmal vorzunehmen.
Beim Glätten von Daten kann man sehr nah beim Original bleiben oder sich aber sehr weit entfernen. Das hängt davon ab, was man für Hintergrundinformationen hat. Also vor allem, wie die Messdaten entstanden sind.
Folgende Fragen ergaben sich für uns:
- Wie schaute die Destillationsvorrichtung aus.
- Womit wurde der Alkoholgehalt gemessen? Über die Dichte (Spindel oder Pyknometer) oder über den Brechungsindex (Refraktometer)? Wenn Refraktometer, wie wurde das Problem des Brechungsindex bei hohen Alkoholstärken gelöst, durch Verdünnung zum Beispiel? Mit welchen Formeln oder Daten wurde aus der Dichte oder dem Brechungsindex die Alkoholstärke ermittelt?
- Nach welchen Daten oder Formeln wurden die Alkoholstärken für den Kessel zusammengemischt.
Und wir haben zu keiner dieser Fragen Antworten. Hätten wir welche, könnten wir abschätzen, wo es eher zu Messfehlern kommt und wo eher nicht. Und dann würden wir die Kurven vielleicht etwas anders zeichnen.
Wir wissen nur, daß diese Daten oft von wissenschaftlichen Publikationen zitiert werden und das sie mit Abstand am meisten Datenpunkte haben verglichen mit anderen Quellen. Es sind 125 Punkte, also 125 mal mol im Kessel, Siedetemperatur und mol im Destillat.
Ein Vergleich mit Computersimulationen wie Aspen Plus verlief enttäuschend. Entweder sind die Messdaten also extrem schlecht oder man kann ein Siedediagramm halt nicht so genau vorhersagen, wie wir es gerne hätten.
Wir denken jedenfalls, daß bei den Messungen so große Fehler nicht passieren können, welche nötig wären, um auf die Werte von Aspen zu kommen, und halten deswegen zu den Messwerten.
Hier ein Bild mit den Originaldaten in rot und unseren daraus gebildeten Kurven in grün:
Es mag an manchen Stellen seltsam sein, wie wir uns von den Messdaten entfernen. In der Mitte der Kurve A zum Beispiel. Dazu sei gesagt, daß diese Stellen ihren Grund in der seltsamen Wellenform der Originaldaten rechts unten Kurve B haben. Wenn man diese korrigiert, verändern sich auch die anderen Kurven, da ja alles zusammenhängt. Bei der Glättung dieser Welle haben versucht, die dabei zwangsläufig entstehenden Abweichung auf die beiden anderen Kurven gleichmäßig zu verteilen.
Es ist allerdings nicht auszuschließen, daß diese Welle physikalische Ursachen hat, es also ein Fehler ist, sie zu glätten. Es gab unterschiedliche Meinungen darüber innerhalb des Rechnerteams.
Die nun richtigere Umrechnung von mol% in vol% hat vor allem bewirkt, daß wir nun der Meinung sind, daß das Azeotrop nicht bei 97.18 sondern bei 97.2vol% liegt.
Die Kurven A und B gehen rechts unten nun etwas spitzer aufeinander zu, was dazu führt, daß laut unserer Rechner nun für sehr hohe Alkoholstärken, also über 95vol%, mehr Böden bzw Rektifikation nötig sind, als bisher berechnet.
Den anfänglichen Anstieg Kurve C links unten haben wir etwas steiler gemacht, weil Daten aus anderen Quellen darauf hindeuten. Es ist optisch nicht stark sichtbar, hat aber auf die Rechner bei sehr niedrigen Alkoholstärken im Kessel merkbare Auswirkungen.
Jedenfalls sind diese neuen Daten nun in alle unsere Rechner eingebaut, wodurch die berechneten Ergebnisse jetzt geringfügig anders sind.
Unser Ziel ist es, irgendwann mal eigene Messungen zu machen. Aber das ist ganz ferne Zukunftsmusik.
Das hatten wir zum Anlass genommen, uns die Daten und vor allem die Kurven, welche wir daraus gemacht hatten, also wie wir die Messdaten zu einem vernünftigen Verlauf geglättet hatten, nochmal vorzunehmen.
Beim Glätten von Daten kann man sehr nah beim Original bleiben oder sich aber sehr weit entfernen. Das hängt davon ab, was man für Hintergrundinformationen hat. Also vor allem, wie die Messdaten entstanden sind.
Folgende Fragen ergaben sich für uns:
- Wie schaute die Destillationsvorrichtung aus.
- Womit wurde der Alkoholgehalt gemessen? Über die Dichte (Spindel oder Pyknometer) oder über den Brechungsindex (Refraktometer)? Wenn Refraktometer, wie wurde das Problem des Brechungsindex bei hohen Alkoholstärken gelöst, durch Verdünnung zum Beispiel? Mit welchen Formeln oder Daten wurde aus der Dichte oder dem Brechungsindex die Alkoholstärke ermittelt?
- Nach welchen Daten oder Formeln wurden die Alkoholstärken für den Kessel zusammengemischt.
Und wir haben zu keiner dieser Fragen Antworten. Hätten wir welche, könnten wir abschätzen, wo es eher zu Messfehlern kommt und wo eher nicht. Und dann würden wir die Kurven vielleicht etwas anders zeichnen.
Wir wissen nur, daß diese Daten oft von wissenschaftlichen Publikationen zitiert werden und das sie mit Abstand am meisten Datenpunkte haben verglichen mit anderen Quellen. Es sind 125 Punkte, also 125 mal mol im Kessel, Siedetemperatur und mol im Destillat.
Ein Vergleich mit Computersimulationen wie Aspen Plus verlief enttäuschend. Entweder sind die Messdaten also extrem schlecht oder man kann ein Siedediagramm halt nicht so genau vorhersagen, wie wir es gerne hätten.
Wir denken jedenfalls, daß bei den Messungen so große Fehler nicht passieren können, welche nötig wären, um auf die Werte von Aspen zu kommen, und halten deswegen zu den Messwerten.
Hier ein Bild mit den Originaldaten in rot und unseren daraus gebildeten Kurven in grün:
Es ist allerdings nicht auszuschließen, daß diese Welle physikalische Ursachen hat, es also ein Fehler ist, sie zu glätten. Es gab unterschiedliche Meinungen darüber innerhalb des Rechnerteams.
Die nun richtigere Umrechnung von mol% in vol% hat vor allem bewirkt, daß wir nun der Meinung sind, daß das Azeotrop nicht bei 97.18 sondern bei 97.2vol% liegt.
Die Kurven A und B gehen rechts unten nun etwas spitzer aufeinander zu, was dazu führt, daß laut unserer Rechner nun für sehr hohe Alkoholstärken, also über 95vol%, mehr Böden bzw Rektifikation nötig sind, als bisher berechnet.
Den anfänglichen Anstieg Kurve C links unten haben wir etwas steiler gemacht, weil Daten aus anderen Quellen darauf hindeuten. Es ist optisch nicht stark sichtbar, hat aber auf die Rechner bei sehr niedrigen Alkoholstärken im Kessel merkbare Auswirkungen.
Jedenfalls sind diese neuen Daten nun in alle unsere Rechner eingebaut, wodurch die berechneten Ergebnisse jetzt geringfügig anders sind.
Unser Ziel ist es, irgendwann mal eigene Messungen zu machen. Aber das ist ganz ferne Zukunftsmusik.
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Re: Pyknometer
Drei neue Rechner sind da.
Es geht um Dichtemessungen mit Pyknometern.
Pyknometer sind sehr praktische und exakte Messinstrumente für die Dichte von Flüssigkeiten und somit für den Zucker- oder Alkoholgehalt:
Pyknometer-Alkoholstärke
Pyknometer-Zuckerkonzentration
Ein Pyknometer ist eigentlich einfach nur ein sehr exaktes und exakt befüllbares Volumen. Man befüllt den Kolben mit der Flüssigkeit, setzt den Stopfen auf und durch die Kapillare im Stopfen entweicht nach oben überschüssige Flüssigkeit, welche abgewischt werden muss. Dann wird das Pyknometer gewogen. Aus dem Gewicht des gefüllten und des leeren Pyknometers und dessen Innenvolumen wird die Dichte berechnet. Die Gewichtsdifferenz geteilt durch das Volumen ergibt die Dichte.
Wenn man eine höhere Genauigkeit haben möchte als mit herkömmlichen Spindeln oder Refraktometern, sind die Formeln, um aus den Messwerten die Dichte zu berechnen, allerdings doch wesentlich komplexer. Genau dafür sind unsere neuen Rechner da. Zudem können auch Messergebnisse bei von 20°C abweichenden Temperaturen eingegeben werden. Diese werden automatisch korrigiert.
Dann braucht man aber auch eine sehr feine Waage und das Pyknometer muss gut in deren Messbereich passen. Gut zum Beispiel ist die Kombination eines Pyknometers mit 50ml Volumen und einer Waage, welche von 1mg bis 100g misst. Oder ein 25ml-Pyknometer mit einer Waage von 1mg-50g.
Die billigstmögliche Lösung, welche aber schon den Anspruch hat, wesentlich genauere Ergebnisse zu liefern als unsere üblichen Messgeräte, wäre sowas:
https://www.amazon.de/dp/B082SSTFLP/ref ... B082SSTFLP
und sowas:
https://www.ebay.de/itm/100g-0-001g-Sch ... 22nhKEgCsg
Das vielleicht größte Problem bei solchen Billigwaagen ist die statische Aufladung des Pyknometers, welche hier nicht von dem Plastikgehäuse abgeleitet wird. Man sollte also zum Beispiel auf keinen Fall das Pyknometer vor dem Messen mit einem Synthetiktuch trockenreiben. Antistatik-Handschuhe sind überlegenswert.
Ehrlich gesagt habe wir aber noch zu wenig Erfahrung mit Feinwaagen. Wir fangen gerade an, uns mit alten, aber sehr hochwertigen, mechanischen Feinwaagen der Firma Mettler zu beschäftigen. Wir müssen erst schauen, wie groß mit etwas Erfahrung der Unterschied zu den elektronischen Billigwaagen ist.
Eine wichtige Sache haben wir aber herausgefunden: Es sind keine genauen und damit teuren Kalibriergewichte nötig. Diesen billigen elektronischen Waagen ist normalerweise ein Kalibriergewicht sehr niedriger Qualität beigegeben. Ein Kalibriergewicht ist auch nötig. Allerdings muss es nicht genau sein, wenn man sowohl bei der Volumenbestimmung als auch später immer die gleiche Waage mit demselben Kalibriergewicht verwendet. Die durch ein ungenaues Kalibriergewicht entstehenden Fehler bei der Volumenbestimmung und dann bei den Dichtebestimmungen heben sich nämlich gegenseitig exakt auf.
Deswegen und weil billige Pyknometer kein genaues Volumen eingraviert haben, haben wir auch ein Tool zur Volumenbestimmung mit einer Wasserwägung:
Pyknometer-Volumenbestimmung
Wir haben die Rechner eher wissenschaftlich ausgelegt: Zum Beispiel Konstanten wie der Volumenausdehnungskoeffizient des Glases oder die Dichte der Luft können abgeändert werden. Es macht die Rechner vielleicht unübersichtlich, aber sie sollen auch für höchstmögliche Präzision verwendbar sein, auch wenn vielleicht nur wenige die entsprechende Waage dafür haben, wo das dann interessant wird.
Übrigens werden auch höhere Erfordernisse an die Temperaturmessung nötig, je genauer man es haben möchte. Das ist bei Spindeln und Refraktometern aber auch so.
Es geht um Dichtemessungen mit Pyknometern.
Pyknometer sind sehr praktische und exakte Messinstrumente für die Dichte von Flüssigkeiten und somit für den Zucker- oder Alkoholgehalt:
Pyknometer-Alkoholstärke
Pyknometer-Zuckerkonzentration
- Pyknometer.jpg (9.59 KiB) 5339 mal betrachtet
Wenn man eine höhere Genauigkeit haben möchte als mit herkömmlichen Spindeln oder Refraktometern, sind die Formeln, um aus den Messwerten die Dichte zu berechnen, allerdings doch wesentlich komplexer. Genau dafür sind unsere neuen Rechner da. Zudem können auch Messergebnisse bei von 20°C abweichenden Temperaturen eingegeben werden. Diese werden automatisch korrigiert.
Dann braucht man aber auch eine sehr feine Waage und das Pyknometer muss gut in deren Messbereich passen. Gut zum Beispiel ist die Kombination eines Pyknometers mit 50ml Volumen und einer Waage, welche von 1mg bis 100g misst. Oder ein 25ml-Pyknometer mit einer Waage von 1mg-50g.
Die billigstmögliche Lösung, welche aber schon den Anspruch hat, wesentlich genauere Ergebnisse zu liefern als unsere üblichen Messgeräte, wäre sowas:
https://www.amazon.de/dp/B082SSTFLP/ref ... B082SSTFLP
und sowas:
https://www.ebay.de/itm/100g-0-001g-Sch ... 22nhKEgCsg
Das vielleicht größte Problem bei solchen Billigwaagen ist die statische Aufladung des Pyknometers, welche hier nicht von dem Plastikgehäuse abgeleitet wird. Man sollte also zum Beispiel auf keinen Fall das Pyknometer vor dem Messen mit einem Synthetiktuch trockenreiben. Antistatik-Handschuhe sind überlegenswert.
Ehrlich gesagt habe wir aber noch zu wenig Erfahrung mit Feinwaagen. Wir fangen gerade an, uns mit alten, aber sehr hochwertigen, mechanischen Feinwaagen der Firma Mettler zu beschäftigen. Wir müssen erst schauen, wie groß mit etwas Erfahrung der Unterschied zu den elektronischen Billigwaagen ist.
Eine wichtige Sache haben wir aber herausgefunden: Es sind keine genauen und damit teuren Kalibriergewichte nötig. Diesen billigen elektronischen Waagen ist normalerweise ein Kalibriergewicht sehr niedriger Qualität beigegeben. Ein Kalibriergewicht ist auch nötig. Allerdings muss es nicht genau sein, wenn man sowohl bei der Volumenbestimmung als auch später immer die gleiche Waage mit demselben Kalibriergewicht verwendet. Die durch ein ungenaues Kalibriergewicht entstehenden Fehler bei der Volumenbestimmung und dann bei den Dichtebestimmungen heben sich nämlich gegenseitig exakt auf.
Deswegen und weil billige Pyknometer kein genaues Volumen eingraviert haben, haben wir auch ein Tool zur Volumenbestimmung mit einer Wasserwägung:
Pyknometer-Volumenbestimmung
Wir haben die Rechner eher wissenschaftlich ausgelegt: Zum Beispiel Konstanten wie der Volumenausdehnungskoeffizient des Glases oder die Dichte der Luft können abgeändert werden. Es macht die Rechner vielleicht unübersichtlich, aber sie sollen auch für höchstmögliche Präzision verwendbar sein, auch wenn vielleicht nur wenige die entsprechende Waage dafür haben, wo das dann interessant wird.
Übrigens werden auch höhere Erfordernisse an die Temperaturmessung nötig, je genauer man es haben möchte. Das ist bei Spindeln und Refraktometern aber auch so.
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Glasthermometer Fadenkorrektur
Wir hatten die Überlegung, ob an unseren Destillen nicht vielleicht größer angelegte Glasthermometer besser sind als unsere elektronischen Sensoren. Es gibt sehr lange Glasthermometer, bei denen man sehr gut auf 0.1°C ablesen kann. Solche haben wir gekauft und damit etwas rumprobiert. Es sind dann aber vorher unterschätzte Probleme aufgetaucht. Das wichtigste davon ist der sogenannte Fadenfehler.
Dafür haben wir einen kleinen Rechner entwickelt:
Glasthermometer-Fadenkorrektur
Er berechnet die sogenannte "Fadenkorrektur".
Die Fadenkorrektur versucht den Einfluß der Eintauchtiefe zu kompensieren. Glasthermometer sind normalerweise "ganz eintauchend" justiert. Also daß der "Faden" (die Flüssigkeit in der Kapillare) die gleiche Temperatur hat wie die Kugel ganz unten.
Dies ist aber bei unseren Destillen nicht möglich. Daher ist der Faden kälter und das Thermometer zeigt immer eine etwas zu niedrige Temperatur an.
Ein Beispiel:
Ein Quecksilberthermometer, welches 90°C anzeigt, dabei bis zum Skalenstrich bei 0°C in den Dampf eingetaucht ist und dessen herausragender Faden im Mittel 40°C hat, zeigt 0.8°C zu wenig an. Wenn es ein Thermometer mit Petroleumfüllung ist, beträgt der Fehler allerdings stolze 4.1°C. Ein Quecksilberthermometer ist also schon mal grundsätzlich wesentlich besser als eines mit Petroleumfüllung. Die Thermometer bis 100 oder 150°C mit farbiger Füllung haben normalerweise eine solche Petroleumfüllung. Das bessere Abschneiden des Quecksilberthermometers liegt daran, daß das Volumen von Quecksilber viel weniger auf Temperaturveränderungen reagiert wie das von Petroleum. Das bedeutet, wenn man zwei Thermometer entwickelt, eines mit Quecksilber, eines mit Petroleum, aber sonst identisch (gleicher Messbereich, gleiche Länge, also Strichabstand), muss man beim Quecksilberthermometer eine viel größere Kugel dranmachen, also viel mehr Flüssigkeit verwenden. Und das hat zur Folge, daß der aus dem Destillationsdampf heraushängende Faden viel kleiner im Vergleich zum Gesamtvolumen ist, also nur ein viel kleinerer Anteil der Flüssigkeit der kühleren Temperatur außerhalb der Destille ausgesetzt ist.
Das schwierige dabei ist, die mittlere Fadentemperatur zu messen. Am Einfachsten geht das, indem man ein zweites Thermometer an das Hauptthermometer klemmt. Und zwar so, daß die Kugel bzw der Sensor bei der Mitte des heraushängenden Fadens des Hauptthermomters liegt. Es gibt aber auch noch andere, weitaus aufwändigere Methoden mit sogenannten "auxiliary stems" (den deutschen Begriff dafür habe ich nicht gefunden) oder mit sogenannten "Fadenthermometern".
Wer sich darin vertiefen will, dem sei diese pdf ans Herz gelegt:
THE CORRECTION FOR EMERGENT STEM OF THE MERCURIAL THERMOMETER
Es gibt auch Thermometer, die für eine bestimmte Eintauchtiefe justiert sind. Oft werden sie zusammen mit einer Laborglas-Destillierbrücke und einem Silikonstopfen verkauft. Bei diesen sind die physikalischen Gesetze natürlich aber nicht außer Kraft gesetzt. Die Skala wurde einfach nur korrigiert. Da die mittlere Fadentemperatur allerdings sehr von der Außentemperatur abhängig ist, und diese je nach Raumtemperatur und auch je nach Isolierung der Destille sehr unterschiedlich sein kann, ist das nicht sehr genau. Wenn es dann noch ein Petroleumthermometer ist, macht das dann auch viel aus. Und vor allem ist es dann auch anders als bei der vorigen Destillation, wenn sich die Außentemperatur geändert hat. Man kann also nicht merh gut mit älteren Messwerten vergleichen.
Mein persönliches Fazit ist, daß ich doch lieber bei den elektronischen Thermometern bleibe.
Wenn jemand von euch mit Glasthermometern liebäugelt, bitte nehmt eines mit Quecksilberfüllung.
Dafür haben wir einen kleinen Rechner entwickelt:
Glasthermometer-Fadenkorrektur
Er berechnet die sogenannte "Fadenkorrektur".
Die Fadenkorrektur versucht den Einfluß der Eintauchtiefe zu kompensieren. Glasthermometer sind normalerweise "ganz eintauchend" justiert. Also daß der "Faden" (die Flüssigkeit in der Kapillare) die gleiche Temperatur hat wie die Kugel ganz unten.
Dies ist aber bei unseren Destillen nicht möglich. Daher ist der Faden kälter und das Thermometer zeigt immer eine etwas zu niedrige Temperatur an.
Ein Beispiel:
Ein Quecksilberthermometer, welches 90°C anzeigt, dabei bis zum Skalenstrich bei 0°C in den Dampf eingetaucht ist und dessen herausragender Faden im Mittel 40°C hat, zeigt 0.8°C zu wenig an. Wenn es ein Thermometer mit Petroleumfüllung ist, beträgt der Fehler allerdings stolze 4.1°C. Ein Quecksilberthermometer ist also schon mal grundsätzlich wesentlich besser als eines mit Petroleumfüllung. Die Thermometer bis 100 oder 150°C mit farbiger Füllung haben normalerweise eine solche Petroleumfüllung. Das bessere Abschneiden des Quecksilberthermometers liegt daran, daß das Volumen von Quecksilber viel weniger auf Temperaturveränderungen reagiert wie das von Petroleum. Das bedeutet, wenn man zwei Thermometer entwickelt, eines mit Quecksilber, eines mit Petroleum, aber sonst identisch (gleicher Messbereich, gleiche Länge, also Strichabstand), muss man beim Quecksilberthermometer eine viel größere Kugel dranmachen, also viel mehr Flüssigkeit verwenden. Und das hat zur Folge, daß der aus dem Destillationsdampf heraushängende Faden viel kleiner im Vergleich zum Gesamtvolumen ist, also nur ein viel kleinerer Anteil der Flüssigkeit der kühleren Temperatur außerhalb der Destille ausgesetzt ist.
Das schwierige dabei ist, die mittlere Fadentemperatur zu messen. Am Einfachsten geht das, indem man ein zweites Thermometer an das Hauptthermometer klemmt. Und zwar so, daß die Kugel bzw der Sensor bei der Mitte des heraushängenden Fadens des Hauptthermomters liegt. Es gibt aber auch noch andere, weitaus aufwändigere Methoden mit sogenannten "auxiliary stems" (den deutschen Begriff dafür habe ich nicht gefunden) oder mit sogenannten "Fadenthermometern".
Wer sich darin vertiefen will, dem sei diese pdf ans Herz gelegt:
THE CORRECTION FOR EMERGENT STEM OF THE MERCURIAL THERMOMETER
Es gibt auch Thermometer, die für eine bestimmte Eintauchtiefe justiert sind. Oft werden sie zusammen mit einer Laborglas-Destillierbrücke und einem Silikonstopfen verkauft. Bei diesen sind die physikalischen Gesetze natürlich aber nicht außer Kraft gesetzt. Die Skala wurde einfach nur korrigiert. Da die mittlere Fadentemperatur allerdings sehr von der Außentemperatur abhängig ist, und diese je nach Raumtemperatur und auch je nach Isolierung der Destille sehr unterschiedlich sein kann, ist das nicht sehr genau. Wenn es dann noch ein Petroleumthermometer ist, macht das dann auch viel aus. Und vor allem ist es dann auch anders als bei der vorigen Destillation, wenn sich die Außentemperatur geändert hat. Man kann also nicht merh gut mit älteren Messwerten vergleichen.
Mein persönliches Fazit ist, daß ich doch lieber bei den elektronischen Thermometern bleibe.
Wenn jemand von euch mit Glasthermometern liebäugelt, bitte nehmt eines mit Quecksilberfüllung.
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Begleitstoffesimulator 1
Ein neuer Rechner ist da. Der Begleitstoffesimulator 1.
Er simuliert das Verhalten wichtiger Aromastoffe während der Destillation. Es ist das größte Projekt bisher. Schön, daß was daraus geworden ist. Groß wird nun auch der Beitrag dazu.
Wir haben von über 50 schnapstypischen Aromastoffen Flüchtigkeitsdaten gefunden.
Die Flüchtigkeit eines Stoffes ist definiert, wie groß der Anteil im Dampf im Vergleich zum Anteil in der Flüssigkeit ist. Eine Flüchtigkeit von 1 bedeutet also, im Dampf ist ein genauso großer Anteil dieses Stoffs wie in der Flüssigkeit.
Von vielen Stoffen haben wir mehrere Quellen gefunden, sodaß wir anhand der Übereinstimmung mit anderen Quellen deren Qualität einschätzen können. Einige Daten haben wir deswegen nicht verwendet.
Leider fehlen ein paar nicht unwichtige Daten: Vor allem Dimethylsulfid, Ethylcarbamat, Ethylbutyrat und Milchsäure hätten wir noch gerne gehabt.
Die Flüchtigkeit von Aromastoffen ist in Wasser immer wesentlich höher als in Alkohol. Alkohol hält die Aromastoffe vom Verdampfen ab. Das merkt man auch daran, daß unverdünnte Schnäpse immer viel weniger riechen als verdünnte. Und wenn manchmal empfohlen wird, Whisky ein paar Tropfen Wasser hinzuzufügen, bevor man ihn trinkt, hat das da seinen Ursprung.
Zwischen der höheren Flüchtigkeit in Wasser und der niedrigeren in Alkohol hängt die Kurve immer durch. Also eine typische Kurve der Flüchtigkeit (y-Achse) in Abhängigkeit zu den vol% (x-Achse) schaut so aus:
Obwohl diese Kurven also alle ähnlich sind, ergibt sich doch eine große Bandbreite an Verhaltensweisen, vor allem je nach dem, wo die Endpunkte der Kurve liegen, aber auch, wie stark die Kurve durchhängt.
Auch die Kurve von Ethanol schaut so aus. Die Flüchtigkeit ist bei (fast) 0vol% ca. 11 und beim Azeotrop 1.
Für unsere Überlegungen ist meist der Verlauf der relativen Flüchtigkeit zu der von Ethanol interessant. An ihm kann man das sehr unterschiedliche Verhalten der Stoffe erkennen. Also z.B. ob sie im Vorlauf oder Nachlauf landen.
Alleine anhand der Kurven, also ohne Simulation, kann man aber nur sehen, wie an einem bestimmten Startpunkt einer Destillation sich die Begleitstoffe verhalten werden. Nicht gut kann man spätere Zeitpunkte vorhersagen, da es schwer einzuschätzen ist, wie viel bei einem späteren Zeitpunkt bei dann höherer Flüchtigkeit, da der Alkoholgehalt gesunken ist, noch von dem Stoff in der Destille ist. Also einerseits hat man dann die höhere Flüchtigkeit andererseits aber nicht mehr den gleichen Anteil in der Destille. Es kann also durchaus sein, daß, obwohl die Flüchtigkeit steigt, immer weniger des Stoffs ins Destillat kommt. Bei allen typischen Vorlaufstoffen ist das so. Typische Vorlaufstoffe sind die, welche über den ganzen Bereich von 0-100vol% eine höhere Flüchtigkeit als Ethanol haben. Typische Nachlaufstoffe haben über den ganzen Bereich eine niedrigere Flüchtigkeit als Ethanol. Und Stoffe mit niedrigerer Flüchtigkeit als Wasser bleiben in der Destille zurück.
Die Destillation muss also in Kleinstschritten durchgerechnet werden. Und das kann nur ein Rechenprogramm.
Während der Arbeit an diesem Simulator ist uns einerseits dessen großes Potential aufgefallen, grundsätzliche Theorien zu bestätigen, zu widerlegen oder zu konkretisieren. Andererseits gibt es aber oft Unterschiede zwischen Theorie und Praxis, z.B. bei hoher Rektifikation. Über die Unterschiede zwischen Theorie und Praxis steht noch einiges in der Anmerkung direkt im Begleitstoffesimulator.
Hier möchte ich noch ein paar Beispiele zeigen, was der Simulator uns generell sagen kann:
Und das sind ja wichtige Fragen, auf die es oft nur vage Antworten gibt.
"From pot stills to continuous stills: flavor modification by distillation" von Robert Piggot. Veröffentlicht in "The Alcohol Textbook", Kapitel 17
"Whisky Technology, Production and Marketing", original von Panek und Boucher, wahrscheinlich "Continuous distillation", veröffentlicht wahrscheinlich auch in "The Science and Technology of Whiskies" von J.R.Piggott, Longman Scientific and Technical
"Chemical aspects of distilling wines into brandy" von J.F. Guymon, 1973, veröffentlicht in "Chemistry of Winemaking"; Webb, A.; Advances in Chemistry; American Chemical Society: Washington, DC, 1974
"Computer simulation applied to studying continuous spirit distillation and product quality control" von Fabio R.M. Batista, Antonio J.A. Meirelles, Food Control 22 (2011) 1592-1603
"Vapor-liquid equilibria of organic homologues in ethanol-water solutions" von C.Williams
"A quick method for obtaining partition factor of congeners in spirits" von A.Martin
"Vapor-Liquid Equilibria Measurements of Bitter Orange Aroma Compounds Highly Diluted in Boiling Hydro-Alcoholic Solutions at 101.3 kPa" von S.Deterre
"Distillation principles and processes" von S.Young, S.308
"Vapor−Liquid Equilibrium of Ethyl Lactate Highly Diluted in Ethanol−Water Mixtures at 101.3 kPa. Experimental Measurements and Thermodynamic Modeling Using Semiempirical Models" von C.Puentes
"Review and thermodynamic modeling with NRTL model of vapor–liquid equilibria (VLE) of aroma compounds highly diluted in ethanol–water mixtures at 101.3 kPa" von C.Puentes
"Modélisation des équilibres entre phases et simulation de la distillation des eaux-de-vie en vue d’une meilleure compréhension du comportement des composés volatils d’arôme" von C.Puentes
"Vapour–liquid equilibria of aroma compounds in hydroalcoholic solutions: Measurements with a recirculation method and modelling with the NRTL and COSMO-SAC approaches" von V.Athes
"Vapor-Liquid Equilibria of a Minute Amount of n-Propyl, Isobutyl, and Isoamyl Alcohols in Aqueous Ethanol Solution under Reduced Pressure" von A.Ikari
"Vapor-Liquid Equilibria of a Minute Amount of Methanol, Isovaleraldehyde and Diacetyl in Aqueous Ethanol Solution under Reduced Pressure" von A.Ikari
"Behavior of a minute amount of furfural in distillation of aqueous ethanol solution under reduced pressure" von A.Ikari
"Vapor-Liquid Equilibria of Trace Isobutyraldehyde, Ethyl Acetate and Isoamyl Acetate in Aqueous Ethanol Solution under Reduced Pressure" von A.Ikari
"Review and thermodynamic modeling with NRTL model of vapor–liquid equilibria (VLE) of aroma compounds highly diluted in ethanol–water mixtures at 101.3 kPa" von C.Puentes
Die Reihenfolge ist zufällig.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
Er simuliert das Verhalten wichtiger Aromastoffe während der Destillation. Es ist das größte Projekt bisher. Schön, daß was daraus geworden ist. Groß wird nun auch der Beitrag dazu.
Wir haben von über 50 schnapstypischen Aromastoffen Flüchtigkeitsdaten gefunden.
Die Flüchtigkeit eines Stoffes ist definiert, wie groß der Anteil im Dampf im Vergleich zum Anteil in der Flüssigkeit ist. Eine Flüchtigkeit von 1 bedeutet also, im Dampf ist ein genauso großer Anteil dieses Stoffs wie in der Flüssigkeit.
Von vielen Stoffen haben wir mehrere Quellen gefunden, sodaß wir anhand der Übereinstimmung mit anderen Quellen deren Qualität einschätzen können. Einige Daten haben wir deswegen nicht verwendet.
Leider fehlen ein paar nicht unwichtige Daten: Vor allem Dimethylsulfid, Ethylcarbamat, Ethylbutyrat und Milchsäure hätten wir noch gerne gehabt.
Die Flüchtigkeit von Aromastoffen ist in Wasser immer wesentlich höher als in Alkohol. Alkohol hält die Aromastoffe vom Verdampfen ab. Das merkt man auch daran, daß unverdünnte Schnäpse immer viel weniger riechen als verdünnte. Und wenn manchmal empfohlen wird, Whisky ein paar Tropfen Wasser hinzuzufügen, bevor man ihn trinkt, hat das da seinen Ursprung.
Zwischen der höheren Flüchtigkeit in Wasser und der niedrigeren in Alkohol hängt die Kurve immer durch. Also eine typische Kurve der Flüchtigkeit (y-Achse) in Abhängigkeit zu den vol% (x-Achse) schaut so aus:
- Eine unserer Quellen für Isobutanol. Die roten Punkte sind die Messwerte, die grüne Linie die von uns dazu gezogene Linie
- Isobutanol.png (8.1 KiB) 3720 mal betrachtet
Obwohl diese Kurven also alle ähnlich sind, ergibt sich doch eine große Bandbreite an Verhaltensweisen, vor allem je nach dem, wo die Endpunkte der Kurve liegen, aber auch, wie stark die Kurve durchhängt.
Auch die Kurve von Ethanol schaut so aus. Die Flüchtigkeit ist bei (fast) 0vol% ca. 11 und beim Azeotrop 1.
Für unsere Überlegungen ist meist der Verlauf der relativen Flüchtigkeit zu der von Ethanol interessant. An ihm kann man das sehr unterschiedliche Verhalten der Stoffe erkennen. Also z.B. ob sie im Vorlauf oder Nachlauf landen.
Alleine anhand der Kurven, also ohne Simulation, kann man aber nur sehen, wie an einem bestimmten Startpunkt einer Destillation sich die Begleitstoffe verhalten werden. Nicht gut kann man spätere Zeitpunkte vorhersagen, da es schwer einzuschätzen ist, wie viel bei einem späteren Zeitpunkt bei dann höherer Flüchtigkeit, da der Alkoholgehalt gesunken ist, noch von dem Stoff in der Destille ist. Also einerseits hat man dann die höhere Flüchtigkeit andererseits aber nicht mehr den gleichen Anteil in der Destille. Es kann also durchaus sein, daß, obwohl die Flüchtigkeit steigt, immer weniger des Stoffs ins Destillat kommt. Bei allen typischen Vorlaufstoffen ist das so. Typische Vorlaufstoffe sind die, welche über den ganzen Bereich von 0-100vol% eine höhere Flüchtigkeit als Ethanol haben. Typische Nachlaufstoffe haben über den ganzen Bereich eine niedrigere Flüchtigkeit als Ethanol. Und Stoffe mit niedrigerer Flüchtigkeit als Wasser bleiben in der Destille zurück.
Die Destillation muss also in Kleinstschritten durchgerechnet werden. Und das kann nur ein Rechenprogramm.
Während der Arbeit an diesem Simulator ist uns einerseits dessen großes Potential aufgefallen, grundsätzliche Theorien zu bestätigen, zu widerlegen oder zu konkretisieren. Andererseits gibt es aber oft Unterschiede zwischen Theorie und Praxis, z.B. bei hoher Rektifikation. Über die Unterschiede zwischen Theorie und Praxis steht noch einiges in der Anmerkung direkt im Begleitstoffesimulator.
Hier möchte ich noch ein paar Beispiele zeigen, was der Simulator uns generell sagen kann:
Die berühmte Frage nach dem Methanol
Die meisten hier wissen, daß sich Methanol nicht im Vorlauf konzentriert. Hier kann man es nachprüfen:
5vol% in der Destille, 0 Rektifikation:
Die Kurve von Methanol ist unter der von Ethanol. Methanol geht also langsamer ins Destillat als Ethanol. Wenn man ca. 5% (0.0495) des Alkohols als Vorlauf abtrennt, ist da nur 3.2% (0.0319) des Methanols drinnen.
Wenn man diesen Raubrand nach 25% des Destilleninhalts beendet:
Dann hat man 93.9% des Ethanols und 81.5% des Methanols gesammelt. das ist ein Ratio von 81.5/93.9=0.868.
Wenn man dagegen den Raubrand erst nach 33% des Destilleninhalts beendet:
Dann hat man 98.3% des Ethanols und 91.4% des Methanols gesammelt. Das ist ein Ratio von 0.93.
Das längere Sammeln hat also den Methanolanteil erhöht. Allerdings nur in geringem Umfang.
Bzw. in anderer Darstellungsweise:
Alles Destillat, bevor man 13.7% des Destilleninhalts destilliert hat, erniedrigt schlussendlich im Schnapsglas den Methanolanteil, alles danach erhöht ihn wieder. Also Vorlauf abzutrennen erhöht den Methanolanteil etwas. Und weit runterbrennen erhöht den Methanolanteil ebenfalls etwas. Eine geringfügige Reduzierung des Methanols bekommt man in diesem Fall also, wenn man keinen Vorlauf abtrennt und nicht weit herunterbrennt.
Nun mit 40vol%:
Im Endeffekt das gleiche. Nur verhält sich Methanol deutlich ähnlicher zu Ethanol, wodurch Abtrennen des Vor- oder Nachlaufs noch weniger Auswirkungen hat.
Nun mit Rektifikation:
5vol%, Rektifikation 5, Zoom x 10:
Durch die Rektifikation konzentriert sich das Ethanol etwas im Vorlauf. Wenn man 5% (0.05) des Alkohols als Vorlauf abtrennt, ist man trotzdem nur 12.3% (0.123) des Methanols losgeworden.
Nun 40vol%, Rektifikation 5, Zoom x 10:
Hier wird man mit dem Vorlaufschnitt nach 5% des Alkohols 27.8% des Methanols los.
Es ist also nicht wirklich effektiv alles. Man bekommt auch mit einer kleineren Refluxdestille das meiste nicht los. Wenn man nun noch höhere Prozente in die Destille füllt und mehr Rektifikation einsetzt, behauptet der Rechner, kann man das Methanol relativ schnell ziemlich komplett abtrennen.
95vol%, Rektifikation 10, Zoom x 10:
Und so macht es auch die Industrie. Nicht die Schnapsindustrie, sondern die für Industriealkohol.
Wir können das im Prinzip auch. In der Praxis wird es allerdings weniger gut klappen, da die richtigen Vorlaufstoffe (welche zum Teil sehr gute Aromastoffe sind) sich "vordrängeln" werden und dann der Effekt ins Spiel kommt, daß sich die Moleküle der Stoffe sich gegenseitig beeinflussen.
Außerdem entsteht Methanol ja eigentlich nur bei der Vergärung von pektinhaltigem Obst. Und das möchte man ja meist ohne oder mit nur wenig Rektifikation brennen. Die Beispiele hier mit Rektifikation sind also nur praxisrelevant, wenn man aus Obst Neutralalkohol machen möchte.
5vol% in der Destille, 0 Rektifikation:
Wenn man diesen Raubrand nach 25% des Destilleninhalts beendet:
Wenn man dagegen den Raubrand erst nach 33% des Destilleninhalts beendet:
Das längere Sammeln hat also den Methanolanteil erhöht. Allerdings nur in geringem Umfang.
Bzw. in anderer Darstellungsweise:
Nun mit 40vol%:
Nun mit Rektifikation:
5vol%, Rektifikation 5, Zoom x 10:
Nun 40vol%, Rektifikation 5, Zoom x 10:
Es ist also nicht wirklich effektiv alles. Man bekommt auch mit einer kleineren Refluxdestille das meiste nicht los. Wenn man nun noch höhere Prozente in die Destille füllt und mehr Rektifikation einsetzt, behauptet der Rechner, kann man das Methanol relativ schnell ziemlich komplett abtrennen.
95vol%, Rektifikation 10, Zoom x 10:
Wir können das im Prinzip auch. In der Praxis wird es allerdings weniger gut klappen, da die richtigen Vorlaufstoffe (welche zum Teil sehr gute Aromastoffe sind) sich "vordrängeln" werden und dann der Effekt ins Spiel kommt, daß sich die Moleküle der Stoffe sich gegenseitig beeinflussen.
Außerdem entsteht Methanol ja eigentlich nur bei der Vergärung von pektinhaltigem Obst. Und das möchte man ja meist ohne oder mit nur wenig Rektifikation brennen. Die Beispiele hier mit Rektifikation sind also nur praxisrelevant, wenn man aus Obst Neutralalkohol machen möchte.
Sind die höheren Alkohole im Nachlauf?
Höhere Alkohole (oft auch Fuselalkohole genannt) werden meist als Nachlaufstoffe bezeichnet. Das ist aber nur teilweise richtig.
5vol%, 0 Rektifikation, kein Zoom:
Die typischen und meisten höheren Alkohol gehen alle schneller ins Destillat über als Ethanol. Es gibt dadurch eine geringfügige Anreicherung im Vorlauf.
Andere Darstellung:
Anfangs bekommt man verglichen mit Ethanol mehr der höheren Alkohole ins Destillat, nach ca. 7% der liter weniger.
40vol%, 0 Rektifikation, kein Zoom:
Einige Kurven sind ein wenig unterhalb der von Ethanol, andere ein wenig über Ethanol. Es gibt also kaum Konzentrierungen oder Zurückhaltungen.
Andere Darstellung:
Hier sieht man dasselbe.
Der Abfall der Kurven rechts kommt daher, daß sich die Stoffe insgesamt etwas schneller aufgebraucht haben als Ethanol. Bei dem Abfall der Kurve ist die Konzentartion der Stoffe in der Destille fast 0, aber Ethanol ist noch vorhanden.
Nun mit Rektifikation:
5vol%, 5 Rektifikation, Zoom x 10:
Die typischen und meisten höheren Alkohole werden im Kessel zurückgehalten und schießen, sobald der Alkoholgehalt im Kessel gesunken ist, steil nach oben. Die Rektifikation reduziert also, daß höhere Akohole in den Vor- und Mittellauf gelangen.
Andere Darstellung:
Hier sieht man exemplarisch an der roten Kurve, daß die aktuelle Menge des höheren Alkohols, kurz bevor der Alkoholgehalt im Dampf absackt, in die Höhe schießt. Daß sie danach gleich wieder abfällt, liegt daran, daß der Stoff sich aufgebraucht hat, also innerhalb kürzester Zeit fast komplett im Destillat gelandet ist.
40vol%, 5 Rektifikation, kein Zoom:
Das gleiche Verhalten, nur etwas extremer. Und der Zeitpunkt, an dem der Ethanolgehalt absackt, ist natürlich später.
Mit höherer Rektifikation kann man die höheren Alkohole noch wesentlich stärker zurückhalten, sowohl in der Theorie als auch in der Praxis. Das hochschießen der höheren Alkohole, sobald das Ethanol rüberdestilliert ist, ist aber nicht so extrem, wie es der Simulator berechnet, da sich die Begleitstoffmoleküle gegenseitig beeinflussen, es also quasi einen "Stau" gibt.
Fazit: Daß die höheren Alkohole Nachlaufstoffe sind, stimmt also nur für Refluxdestillen.
5vol%, 0 Rektifikation, kein Zoom:
Andere Darstellung:
40vol%, 0 Rektifikation, kein Zoom:
Andere Darstellung:
Der Abfall der Kurven rechts kommt daher, daß sich die Stoffe insgesamt etwas schneller aufgebraucht haben als Ethanol. Bei dem Abfall der Kurve ist die Konzentartion der Stoffe in der Destille fast 0, aber Ethanol ist noch vorhanden.
Nun mit Rektifikation:
5vol%, 5 Rektifikation, Zoom x 10:
Andere Darstellung:
40vol%, 5 Rektifikation, kein Zoom:
Mit höherer Rektifikation kann man die höheren Alkohole noch wesentlich stärker zurückhalten, sowohl in der Theorie als auch in der Praxis. Das hochschießen der höheren Alkohole, sobald das Ethanol rüberdestilliert ist, ist aber nicht so extrem, wie es der Simulator berechnet, da sich die Begleitstoffmoleküle gegenseitig beeinflussen, es also quasi einen "Stau" gibt.
Fazit: Daß die höheren Alkohole Nachlaufstoffe sind, stimmt also nur für Refluxdestillen.
Vorlauf abtrennen auch beim Raubrand?
Dies ist eine bisher unbeantwortete Frage. Natürlich gibt es Meinungen, aber diese sind bisher durch nichts untermauert.
Zuerst die Frage, was effizienter ist, also ob man beim Rau- oder beim Feinbrand den Vorlauf effizienter abtrennen kann. Die wichtigsten Vorlaufstoffe sind Acetaldehyd und Ethylacetat:
5vol%, 0.3 Rektifikation, Zoom x 10:
Wenn man 5% des Ethanols als Vorlauf abtrennt, hat man 51.9% des Acetaldehyds und 74% des Ethylacetats entfernt.
Nun 40vol%:
Wenn man 5% des Ethanols als Vorlauf abtrennt, hat man 54.5% des Acetaldehyds (also etwa gleich viel wie bei 5vol%) und 36.7% (also nur die Hälfte) des Ethylacetats entfernt.
Das Entfernen des Vorlaufs ist also beim Raubrand effizienter als beim Feinbrand.
Zwei Punkte relativieren dieses Ergebnis allerdings:
- Nach dem Raubrand kann neuer Vorlauf entstehen. Also ein Abtrennen beim Feinbrand muss meist trotzdem gemacht werden.
- Wird der Feinbrand mit einer Refluxdestille gemacht, ist das Abtrennen natürlich wesentlich effizienter:
40vol%, 5 Rektifikation, Zoom x 10:
Schon nach 1% des Ethanols sind beide Vorlaufstoffe so gut wie vollständig entfernt. Ob das wirklich ganz so komplett und schnell auch in der Praxis passiert, ist allerdings nicht sicher. Aber am allgemeinen Ergebnis ändert das nichts.
Meist geht es aber nicht darum, möglichst wenig Ethanol beim Vorlaufabtrennen zu verlieren, sondern man möchte das geschmacklich beste Ergebnis haben. Man möchte beim Vorlaufabtrennen möglichst viele schlechte Aromen abtrennen, aber möglichst ohne, daß dabei auch viele gute Aromen abgetrennt werden.
Und bei dieser Frage kann der Simulator durch seine vergleichende Darstellungen Antworten liefern, indem er darstellt, wie sich innerhalb einer Stoffgruppe die eher guten und die eher schlechten Aromastoffe zueinander verhalten. Die eher schlechten Stoffe sind die mit den niedrigen Molmassen und die eher guten die mit hohen Molmassen. Und nach Molmasse sind die Stoffe im Simulator geordnet. Also das schlechteste (und gleichzeitig häufigste) Aldehyd ist Acetalehyd.
5vol%, 0.3 Rektifikation, Zoom x 10 (nur von der Molekularstruktur bzw. Summenformel typische Aldehyde bzw. mit Acetaldehyd vergleichbare Aldehyde sind dargestellt):
Acetaldehyd (siehe Pfeil) konzentriert sich am wenigsten. Beim Vorlaufabtrennen reduziert man also in diesem Fall mehr die guten als die schlechten Aromen.
Andere Darstellung:
Alle Kurven beginnen über der 1, also über der Kurve von Acetaldehyd.
nun 40vol%:
Bei höherem Alkoholgehalt konzentriert sich Acetaldehyd (Pfeil) besser als fast alle anderen (besseren) Aldehyde.
Andere Ansicht:
Fast alle Kurven beginnen unter der 1.
Das klappt aber nur mit etwas Rektifikation (0.3). Rektifikation ist aber bei jeder Destille beim Vorlauf vorhanden. Ohne Rektifikation:
Hier nun sind die meisten Aldehyde leider über Acetaldehyd. Das bedeutet, zumindest bezüglich der Aldehyde ist eine gewisse Menge an Rektifikation beim Vorlauf aromaverbessernd. Die Rektifikation erhöht man durch langames abdestillieren des Vorlaufs. Womit auch eine theoretische Begründung erbracht wäre, warum man den Vorlauf langsam abdestillieren soll.
Bezüglich der Aldehyde ist ein Vorlaufabtrennen beim Raubrand also aromaschädigend.
Nun die Ester:
5vol%, 0.3 Rektifikation, Zoom x 10 (nur von der Molekularstruktur bzw. Summenformel typische Ester bzw. mit Ethylacetat vergleichbare Ester sind dargestellt):
Fast alle Stoffe konzentieren sich mehr im Vorlauf als Ethylacetat (siehe Pfeil). Beim Vorlaufabtrennen reduziert man in diesem Fall also mehr die guten als die schlechten Aromen.
Andere Darstellung:
Alle Kurven beginnen über der 1, also über der Kurve von Ethylacetat.
nun 40vol%, Zoom x 3:
Bei höherem Alkoholgehalt konzentriert sich Ethylacetat (Pfeil) besser als alle anderen (besseren) Ester.
Andere Darstellung:
Alle Kurven beginnen unter der 1.
Bezüglich der Ester ist ein Vorlaufabtrennen beim Raubrand also ebenfalls aromaschädigend.
Zuerst die Frage, was effizienter ist, also ob man beim Rau- oder beim Feinbrand den Vorlauf effizienter abtrennen kann. Die wichtigsten Vorlaufstoffe sind Acetaldehyd und Ethylacetat:
5vol%, 0.3 Rektifikation, Zoom x 10:
Nun 40vol%:
Das Entfernen des Vorlaufs ist also beim Raubrand effizienter als beim Feinbrand.
Zwei Punkte relativieren dieses Ergebnis allerdings:
- Nach dem Raubrand kann neuer Vorlauf entstehen. Also ein Abtrennen beim Feinbrand muss meist trotzdem gemacht werden.
- Wird der Feinbrand mit einer Refluxdestille gemacht, ist das Abtrennen natürlich wesentlich effizienter:
40vol%, 5 Rektifikation, Zoom x 10:
Meist geht es aber nicht darum, möglichst wenig Ethanol beim Vorlaufabtrennen zu verlieren, sondern man möchte das geschmacklich beste Ergebnis haben. Man möchte beim Vorlaufabtrennen möglichst viele schlechte Aromen abtrennen, aber möglichst ohne, daß dabei auch viele gute Aromen abgetrennt werden.
Und bei dieser Frage kann der Simulator durch seine vergleichende Darstellungen Antworten liefern, indem er darstellt, wie sich innerhalb einer Stoffgruppe die eher guten und die eher schlechten Aromastoffe zueinander verhalten. Die eher schlechten Stoffe sind die mit den niedrigen Molmassen und die eher guten die mit hohen Molmassen. Und nach Molmasse sind die Stoffe im Simulator geordnet. Also das schlechteste (und gleichzeitig häufigste) Aldehyd ist Acetalehyd.
5vol%, 0.3 Rektifikation, Zoom x 10 (nur von der Molekularstruktur bzw. Summenformel typische Aldehyde bzw. mit Acetaldehyd vergleichbare Aldehyde sind dargestellt):
Andere Darstellung:
nun 40vol%:
Andere Ansicht:
Das klappt aber nur mit etwas Rektifikation (0.3). Rektifikation ist aber bei jeder Destille beim Vorlauf vorhanden. Ohne Rektifikation:
Bezüglich der Aldehyde ist ein Vorlaufabtrennen beim Raubrand also aromaschädigend.
Nun die Ester:
5vol%, 0.3 Rektifikation, Zoom x 10 (nur von der Molekularstruktur bzw. Summenformel typische Ester bzw. mit Ethylacetat vergleichbare Ester sind dargestellt):
Andere Darstellung:
nun 40vol%, Zoom x 3:
Andere Darstellung:
Bezüglich der Ester ist ein Vorlaufabtrennen beim Raubrand also ebenfalls aromaschädigend.
Vorlauf bei Aromabränden mit oder ohne Rektifikation abtrennen?
Daß das mit Rektifikation effektiver ist, also daß man so weniger Ethanol verliert, steht außer Frage. Aber kann es vielleicht sein, daß man auf diese Weise mehr von den schlechten Vorlaufstoffen verliert und weniger von den guten? Die Ergebnisse bei dem Vergleich 0 und 0.3 Rektifikation weisen ja darauf hin.
40vol%, 0 Rektifikation, Zoom x 10:
Die Werte unter den Stoffen sind an dem Punkt, wo mindestens 50% sowohl von Acetaldehyd als auch Ethylacetat abgetrennt sind. Man sieht, wie auch von wertvollen Stoffen schon viel ins Destillat, also in den Vorlauf, gekommen ist.
nun mit 5 Rektifikation:
Die Werte sind da, wo die gleiche Menge an Ethylacetat entfernt wurde wie ohne Rektifikation. Schlechte Stoffe wie Acrolein oder Aceton werden nun viel effektiver mit dem Vorlauf abgetrennt. Wertvolle Stoffe wie die großen Aldehyde, die großen Ester und die Zitrusketone und -terpene (und damit wahrscheinlich auch andere wertvolle Ketone und Terpene, von denen wir leider keine Daten haben) in der untersten Reihe rechts werden geschohnt.
Der Übersichtlichkeit halber habe ich die Säuren nicht dargestellt. Denn diese haben mit dem Vorlauf in jedem Fall nichts zu tun.
Ebenfalls nicht dargestellt werden die höheren Alkohole. Ich habe es aber nachgeprüft, diese würden in diesem Fall ohne Rektifikation im Schnitt zu 15% abgetrennt werden und mit Rektifikation so gut wie gar nicht. Hier ist ein kleiner Vorteil des Vorlaufabtrennens ohne Reflux.
Insgesamt ist aber ganz klar ersichtlich, daß ein Vorlaufabtrennen mit Reflux nicht nur effektiver ist sondern auch qualitativ besser.
40vol%, 0 Rektifikation, Zoom x 10:
nun mit 5 Rektifikation:
Der Übersichtlichkeit halber habe ich die Säuren nicht dargestellt. Denn diese haben mit dem Vorlauf in jedem Fall nichts zu tun.
Ebenfalls nicht dargestellt werden die höheren Alkohole. Ich habe es aber nachgeprüft, diese würden in diesem Fall ohne Rektifikation im Schnitt zu 15% abgetrennt werden und mit Rektifikation so gut wie gar nicht. Hier ist ein kleiner Vorteil des Vorlaufabtrennens ohne Reflux.
Insgesamt ist aber ganz klar ersichtlich, daß ein Vorlaufabtrennen mit Reflux nicht nur effektiver ist sondern auch qualitativ besser.
Wie weit den Raubrand herunterbrennen?
Schuld am schechten Geruch und Geschmack von Nachlauf sind vor allem Carbonsäuren. Sie sind etwas, was man eigentlich möglichst wenig im Schnapsglas haben möchte. Andererseits sind sie Vorläufer von Estern. Und je größer die Säuremoleküle, desto besser die Ester. Also auch hier gilt, daß die Säuren, welche im Simulator weiter rechts stehen, die wertvolleren sind. Ganz links die Ameisensäure kommt recht wenig vor, die zweite, die Essigsäure, ist bei weitem die häufigste. Diese beiden linken definiere ich jetzt mal als die schlechten. Die weiter rechts als die guten. Wenn Säuren irgendwann zwischen dem Rau- und Feinbrand oder während des Feinbrands verestern, landen diese Ester größtenteils im Vorlauf und im Mittellauf. Und die Säuren, welche nicht verestern, werden beim Feinbrand dann möglichst vollständig als Nachlauf abgetrennt.
Zuerst eine schwache Maische mit 5vol%, 0 Rektifikaton, kein Zoom:
Das schaut schonmal gut aus. Die Ameisensäure und die Essigsäure (Pfeile) gehen als letzte ins Destillat.
Andere Darstellung:
Hier sieht man nun sehr gut, daß, wenn man sehr lange raubrennt, also bei 5vol% anfangs ist 40% des Kesselinhalts rauzubrennen sehr lange, immer noch mehr bessere Säuren als schlechtere Säuren ins Destillat gehen. Erst je nach Begleitstoff bei zwischen 50 und 70% des Destilleninhalts würde sich das umkehren.
Noch eine andere Darstellung:
Ameisen- und Essigsäure (siehe Pfeile) haben ihren Peak erst ganz am Ende. Die anderen Säuren dagegen je nach Säure bei zwischen 12 und 37% der liter.
Nun eine starke Maische mit 18vol%:
Das schaut ähnlich wie bei 5vol% aus.
Andere Darstellung:
Bei 18vol% im Kessel würde extrem weit runterbrennen vielleicht so bis 60% des Destilleninhalts bedeuten. Alle bis auf eine der "guten" Säuren, sind da noch über 1, gehen also mehr ins Destillat über als die Essigsäure.
Noch eine andere Darstellung:
Die Peaks der guten Säuren sind je nach Säure bei so zwischen 35 und 53% der liter. Ameisen- und Essigsäure (Pfeile) haben den Peak erst ganz am Ende.
Sowohl bei niedrigprozentigen als auch hochprozentigen Maischen scheint ein weit Herunterbrennen bezüglich der Säuren also sinnvoll, zumindest wenn man es auf die Erzeugung von Aromen abzielt. Denn man holt immer anteilig mehr von den besseren als von den schlechteren Säuren ins Destillat.
Andererseits ist aber auch an diesen Diagrammen entnehmbar, daß das Verhältnis von guten zu schlechten Säuren mit der Zeit immer schlechter wird. Also es werden zwar permanent anteilig mehr gute als schlechte Säuren destilliert, aber die schlechten holen auf, da ja die guten in der Destille immer weniger werden und deswegen mit der Zeit immer weniger verdampfen.
Dies in Geschmacksqualität zu übersetzen, ist nicht möglich.
Zusätzliche Faktoren sind, daß dann beim Feinbrand die schlechten Säuren wieder anteilig mehr im Kessel bleiben werden als die guten Säuren und daß die Ester der schlechten Säuren die sein werden, welche sich eher gut im Vorlauf abtrennen lassen werden.
Das sind Hinweise, daß man eher daran denken sollte, die guten Stoffe zu bekommen, als die schlechten loszuwerden. Außer natürlich bei einem Feinbrand.
Zuerst eine schwache Maische mit 5vol%, 0 Rektifikaton, kein Zoom:
Andere Darstellung:
Noch eine andere Darstellung:
Nun eine starke Maische mit 18vol%:
Andere Darstellung:
Noch eine andere Darstellung:
Sowohl bei niedrigprozentigen als auch hochprozentigen Maischen scheint ein weit Herunterbrennen bezüglich der Säuren also sinnvoll, zumindest wenn man es auf die Erzeugung von Aromen abzielt. Denn man holt immer anteilig mehr von den besseren als von den schlechteren Säuren ins Destillat.
Andererseits ist aber auch an diesen Diagrammen entnehmbar, daß das Verhältnis von guten zu schlechten Säuren mit der Zeit immer schlechter wird. Also es werden zwar permanent anteilig mehr gute als schlechte Säuren destilliert, aber die schlechten holen auf, da ja die guten in der Destille immer weniger werden und deswegen mit der Zeit immer weniger verdampfen.
Dies in Geschmacksqualität zu übersetzen, ist nicht möglich.
Zusätzliche Faktoren sind, daß dann beim Feinbrand die schlechten Säuren wieder anteilig mehr im Kessel bleiben werden als die guten Säuren und daß die Ester der schlechten Säuren die sein werden, welche sich eher gut im Vorlauf abtrennen lassen werden.
Das sind Hinweise, daß man eher daran denken sollte, die guten Stoffe zu bekommen, als die schlechten loszuwerden. Außer natürlich bei einem Feinbrand.
Wie weit Nachlauf sammeln?
Dies ist eine ähnliche Frage, wie die davor. Und auch die Antwort ist ähnlich, zumindest wenn man es auf die Erzeugung von Aromen abziehlt.
Ein Feinbrand hat oft so 30vol% im Kessel und der Nachlauf beginnt dann meistens irgendwo bei 10vol% im Kessel.
30vol%, 0 Rektifikation, kein Zoom:
Die senkrechte Linie ist da, wo der Nachlauf ganz gut anfangen könnte. Im Schnitt sind 15.76% der Säuren im Mittellauf gelandet, bei den schlechten sind es 8.95%, bei den guten 18.48%. Das Verhältnis der Säuren gut:schlecht ist im Mittellauf also 2.1.
Nun Nachlauf bis nur 20vol% im aktuellen Dampf, also ein recht kurzes Nachlaufsammeln:
Von den schlechten Säuren sind nun im Schnitt 17.1% gesammelt. Abzüglich der 8.95% des Mittellaufs sind es im Nachlauf also 8.15%.
Die guten Säuren sind zu 39% gesamelt. Abzüglich der 18.48% des Mittellaufs also 20.52%. Das gut:schlecht-Verhältnis der Säuren im Nachlauf ist also 2.5. Der Nachlauf ist also bezüglich dieses Verhältnisses besser als der Mittellauf. Man darf aber ja nicht vergessen, daß diese Säuren an sich nur als Vorläufer für Aromen als wertvoll angesehen werden können. Also diese bessere Verhältnis bedeutet nicht, daß der Nachlauf ein besseres Aroma als der Mittellauf hat, sondern nur, daß diese Säuren das bessere Potential haben.
Nun weiterbrennen bis 1vol% im aktuellen Dampf:
Von den schlechten Säuren sind nun im Schnitt 30.2% gesammelt. Abzüglich der bisherigen 17.1% sind es in diesem Abschnitt (20 - 1vol% im aktuellen Dampf) also 13.1%.
Die guten Säuren sind zu 65.46% gesammelt. Abzüglich der bisherigen 39% also 26.46%.
Das gut:schlecht-Verhältnis der Säuren im zweiten Nachlauf ist also 2.
Das Verhältnis wird also inzwischen schlechter (im ersten Teil des Nachlaufs war es ja 2.5).
Und wieder bedeutet das: Dies in Geschmacksqualität zu übersetzen, ist nicht möglich.
Und ebenfalls sind zusätzliche Faktoren wieder, daß dann bei der Weiterverwendung des Nachlaufs die schlechten Säuren wieder anteilig mehr im Kessel bleiben werden als die guten Säuren und daß die Ester der schlechten Säuren die sein werden, welche sich eher gut im Vorlauf abtrennen lassen werden.
Das sind also ebenfalls Hinweise, daß man beim Nachlaufsammeln eher daran denken sollte, die guten Stoffe zu bekommen, als die schlechten loszuwerden.
Ein Feinbrand hat oft so 30vol% im Kessel und der Nachlauf beginnt dann meistens irgendwo bei 10vol% im Kessel.
30vol%, 0 Rektifikation, kein Zoom:
Nun Nachlauf bis nur 20vol% im aktuellen Dampf, also ein recht kurzes Nachlaufsammeln:
Die guten Säuren sind zu 39% gesamelt. Abzüglich der 18.48% des Mittellaufs also 20.52%. Das gut:schlecht-Verhältnis der Säuren im Nachlauf ist also 2.5. Der Nachlauf ist also bezüglich dieses Verhältnisses besser als der Mittellauf. Man darf aber ja nicht vergessen, daß diese Säuren an sich nur als Vorläufer für Aromen als wertvoll angesehen werden können. Also diese bessere Verhältnis bedeutet nicht, daß der Nachlauf ein besseres Aroma als der Mittellauf hat, sondern nur, daß diese Säuren das bessere Potential haben.
Nun weiterbrennen bis 1vol% im aktuellen Dampf:
Die guten Säuren sind zu 65.46% gesammelt. Abzüglich der bisherigen 39% also 26.46%.
Das gut:schlecht-Verhältnis der Säuren im zweiten Nachlauf ist also 2.
Das Verhältnis wird also inzwischen schlechter (im ersten Teil des Nachlaufs war es ja 2.5).
Und wieder bedeutet das: Dies in Geschmacksqualität zu übersetzen, ist nicht möglich.
Und ebenfalls sind zusätzliche Faktoren wieder, daß dann bei der Weiterverwendung des Nachlaufs die schlechten Säuren wieder anteilig mehr im Kessel bleiben werden als die guten Säuren und daß die Ester der schlechten Säuren die sein werden, welche sich eher gut im Vorlauf abtrennen lassen werden.
Das sind also ebenfalls Hinweise, daß man beim Nachlaufsammeln eher daran denken sollte, die guten Stoffe zu bekommen, als die schlechten loszuwerden.
Gibt es eine wertvolle Fraktion nach dem Nachlauf?
Immer mal wieder liest man, daß nach dem Nachlauf noch eine Fraktion gesammelt wird, welche dann zum Mittellauf gegeben wird. Diese wird oft "sweet water" oder z.B. in Arroyos Schriften über die Rumherstellung "fifth fraction" genannt (foreshots, heads, hearts und tails sind die Fraktionen 1-4). Diese Fraktion soll besser sein als die davor, vor allem süßer, oder vielleicht ist weniger sauer gemeint.
Der große Unterschied zu den vorigen die Säuren betreffende Fragen ist, daß die "guten" (also großen) Säuren hier nicht gut sondern schlecht sind. Denn sie sind hier nicht Vorläufer von Estern und die übrigen unveresterten Säuren werden mit dem Nachlauf abgetrennt, sondern sie sollen hier direkt in den Mittellauf gegeben werden. Und die großen Carbonsäuren haben an sich einen wirklich widerlichen Geruch. Wer die Wahl hat, eine Flasche Essig oder einer Flasche Buttersäure zu verschütten, wird garantiert die Flasche Essig wählen. Ich schreibe deswegen hier nicht mehr von guten und schlechten Säuren, sondern von großen und kleinen.
30vol%, 0 Rektifikation, kein Zoom:
Der Peak der großen Säuren liegt in einem Bereich, wo noch Alkohol im Dampf ist. So etwa bei 10vol% im Dampf. Also bei den meisten im Nachlauf und bei manchen nach dem Nachlauf. Und die kleine Essigsäure steigt langsam aber stetig an. Die y-Achse dieses Diagramms geht vom berechneten Minimalwert bis zum berechneten Maximalwert dieses Stoffes. Jeder Stoff hat also eine andere y-Achse. Essigsäure macht ja normalerweise den Löwenanteil der Säuren aus. Deswegen bedeutet das langsame Ansteigen der Essigsäure eine größere Säuerung des Destillats als das Wegfallen der großen Säuren eine Verringerung der Säuerung bedeutet. Daß das Destillat nach dem Peak weniger sauer wird, kann man also nicht so sagen. Wohl kann man sagen, daß die großen Säuren in der Fraktion nach dem Nachlauf weniger vertreten sind als davor. Also deren penetranter Geruch findet sich vor allem gegen Ende des Nachlaufs wieder, nicht in der Fraktion danach.
Andere Darstellung:
Hier sieht man das Verhältnis zwischen aktueller Essigsäure und den anderen Säuren. Der Abstand wird langsam größer. Bei etwa 50vol% im Dampf, also wahrscheinlich am Anfang des Nachlaufs, ist er am größten, dann nähern sich die Werte an und etwa, wenn man 70% des Destilleninhalts destilliert hat, also in der Fraktion nach dem Nachlauf, kreuzen die Kurven die Linie von Essigsäure. Ab da sammelt man also anteilig weniger große Säuren als Essigsäure.
Nun mal schauen, ob etwas Rektifikation was daran ändert. 0.3 Rektifikation:
Der Peak der großen Säuren ist spitzer und später, bei etwa 2vol% im Dampf. Das ändert aber im Prinzip nicht viel. Nur der Punkt, ab dem die großen Säuren auftauchen, ist später, und der Punkt, ab dem sie wieder verschwinden, ist früher. Im Bezug auf die Fragestellung bedeutet das, daß Rektifikation die mit großen Säuren belastete Fraktion komprimiert, also verkleinert.
Zusammenfassend kann man vielleicht sagen, daß es eine Fraktion nach dem Nachlauf gibt, welche grundsätzlich anders ist als die davor, weil sie nämlich eine Entwicklung zeigt, welche vorher nicht da war: das Verschwinden der großen Säuren. Diese Fraktion ist sehr spät. Allerdings wegen der allgemeinen Regel, daß die Flüchtigkeit der Begleitstoffe in Wasser höher ist als in Alkohol, ist es unmöglich, daß ein Stoff, der beim Feinbrand ganz spät ins Destillat kommt, dann im Glas (also bei Trinkstärke) einen wahrnehmbaren Geruch hat. Die Fraktion nach dem Nachlauf kann dem Schnaps also nur einen von der Nase nicht wahrgenommenen Geschmack oder vielleicht auch nur ein Mundgefühl hinzufügen.
Und Langzeiteffekte, nämlich daß bei der Lagerung Säuren verestern, ist eher ein Argument gegen das Verwenden der Fraktion nach dem Nachlauf. Denn die Essigsäure bildet nicht so interessante Ester wie die größeren Säuren.
Der große Unterschied zu den vorigen die Säuren betreffende Fragen ist, daß die "guten" (also großen) Säuren hier nicht gut sondern schlecht sind. Denn sie sind hier nicht Vorläufer von Estern und die übrigen unveresterten Säuren werden mit dem Nachlauf abgetrennt, sondern sie sollen hier direkt in den Mittellauf gegeben werden. Und die großen Carbonsäuren haben an sich einen wirklich widerlichen Geruch. Wer die Wahl hat, eine Flasche Essig oder einer Flasche Buttersäure zu verschütten, wird garantiert die Flasche Essig wählen. Ich schreibe deswegen hier nicht mehr von guten und schlechten Säuren, sondern von großen und kleinen.
30vol%, 0 Rektifikation, kein Zoom:
Andere Darstellung:
Nun mal schauen, ob etwas Rektifikation was daran ändert. 0.3 Rektifikation:
Zusammenfassend kann man vielleicht sagen, daß es eine Fraktion nach dem Nachlauf gibt, welche grundsätzlich anders ist als die davor, weil sie nämlich eine Entwicklung zeigt, welche vorher nicht da war: das Verschwinden der großen Säuren. Diese Fraktion ist sehr spät. Allerdings wegen der allgemeinen Regel, daß die Flüchtigkeit der Begleitstoffe in Wasser höher ist als in Alkohol, ist es unmöglich, daß ein Stoff, der beim Feinbrand ganz spät ins Destillat kommt, dann im Glas (also bei Trinkstärke) einen wahrnehmbaren Geruch hat. Die Fraktion nach dem Nachlauf kann dem Schnaps also nur einen von der Nase nicht wahrgenommenen Geschmack oder vielleicht auch nur ein Mundgefühl hinzufügen.
Und Langzeiteffekte, nämlich daß bei der Lagerung Säuren verestern, ist eher ein Argument gegen das Verwenden der Fraktion nach dem Nachlauf. Denn die Essigsäure bildet nicht so interessante Ester wie die größeren Säuren.
Hier die komplette Liste unserer Quellen
"From pot stills to continuous stills: flavor modification by distillation" von Robert Piggot. Veröffentlicht in "The Alcohol Textbook", Kapitel 17
"Whisky Technology, Production and Marketing", original von Panek und Boucher, wahrscheinlich "Continuous distillation", veröffentlicht wahrscheinlich auch in "The Science and Technology of Whiskies" von J.R.Piggott, Longman Scientific and Technical
"Chemical aspects of distilling wines into brandy" von J.F. Guymon, 1973, veröffentlicht in "Chemistry of Winemaking"; Webb, A.; Advances in Chemistry; American Chemical Society: Washington, DC, 1974
"Computer simulation applied to studying continuous spirit distillation and product quality control" von Fabio R.M. Batista, Antonio J.A. Meirelles, Food Control 22 (2011) 1592-1603
"Vapor-liquid equilibria of organic homologues in ethanol-water solutions" von C.Williams
"A quick method for obtaining partition factor of congeners in spirits" von A.Martin
"Vapor-Liquid Equilibria Measurements of Bitter Orange Aroma Compounds Highly Diluted in Boiling Hydro-Alcoholic Solutions at 101.3 kPa" von S.Deterre
"Distillation principles and processes" von S.Young, S.308
"Vapor−Liquid Equilibrium of Ethyl Lactate Highly Diluted in Ethanol−Water Mixtures at 101.3 kPa. Experimental Measurements and Thermodynamic Modeling Using Semiempirical Models" von C.Puentes
"Review and thermodynamic modeling with NRTL model of vapor–liquid equilibria (VLE) of aroma compounds highly diluted in ethanol–water mixtures at 101.3 kPa" von C.Puentes
"Modélisation des équilibres entre phases et simulation de la distillation des eaux-de-vie en vue d’une meilleure compréhension du comportement des composés volatils d’arôme" von C.Puentes
"Vapour–liquid equilibria of aroma compounds in hydroalcoholic solutions: Measurements with a recirculation method and modelling with the NRTL and COSMO-SAC approaches" von V.Athes
"Vapor-Liquid Equilibria of a Minute Amount of n-Propyl, Isobutyl, and Isoamyl Alcohols in Aqueous Ethanol Solution under Reduced Pressure" von A.Ikari
"Vapor-Liquid Equilibria of a Minute Amount of Methanol, Isovaleraldehyde and Diacetyl in Aqueous Ethanol Solution under Reduced Pressure" von A.Ikari
"Behavior of a minute amount of furfural in distillation of aqueous ethanol solution under reduced pressure" von A.Ikari
"Vapor-Liquid Equilibria of Trace Isobutyraldehyde, Ethyl Acetate and Isoamyl Acetate in Aqueous Ethanol Solution under Reduced Pressure" von A.Ikari
"Review and thermodynamic modeling with NRTL model of vapor–liquid equilibria (VLE) of aroma compounds highly diluted in ethanol–water mixtures at 101.3 kPa" von C.Puentes
Die Reihenfolge ist zufällig.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
-
Hüse
- Beiträge: 102
- Registriert: 29. Sep 2020, 15:18
Re: Rechner
Wahnsinn was ihr hier so veranstaltet. Respekt!
Hab mich bevor ich das Hobby für mich entdeckt habe, etwas belesen.
Nur muss ich sagen, hier finde ich Informationen die mir noch nirgens über den Weg gelaufen sind. Hab erlich gesagt den Horizont noch nicht gesehen
Hab ein gutes Gefühl hier dabei sein zu dürfen
Muss definitiv noch bissl lesen. Das Studium hat begonnen
Hab mich bevor ich das Hobby für mich entdeckt habe, etwas belesen.
Nur muss ich sagen, hier finde ich Informationen die mir noch nirgens über den Weg gelaufen sind. Hab erlich gesagt den Horizont noch nicht gesehen
Hab ein gutes Gefühl hier dabei sein zu dürfen
Muss definitiv noch bissl lesen. Das Studium hat begonnen
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
pH-Puffer
Ein neuer Rechner ist da: pH-Puffer
Beziehungsweise sind es mehrere Rechner. Es geht um die Einstellung und Pufferung des pH von Maischen mit Hilfe von Zutaten wie Citronensäure und Natriumhydroxid.
Der Vorteil der Zugabe eines solchen Puffers verglichen mit der Zugabe zum Beispiel nur von Citronensäure oder auch Biogen M ist, daß er nicht nur den pH senkt, sondern auch einen Schutz gegen weiteres Absinken des pHs bietet.
Und der Vorteil gegenüber einer Zugabe zum Beispiel nur von Calciumcarbonat ist, daß man mit einem Puffer von Anfang an den pH gesenkt hat, nicht erst nachdem die Gärung in Schwung gekommen ist.
Alles weitere steht direkt in der Anmerkung des Rechners.
Hier aber noch ein praktisches Beispiel:
Angenommen, man möchte 10 liter Maische zwischen pH 3.5 und 3.2 vergären und den Puffer aus wasserfreie Citronensäure und Natriumydroxid herstellen:
Da man nicht erwartet, daß der pH ohne Puffer unter 3 sinken würde, reicht eine Pufferkapazität von 1 (siehe Anmerkung im Rechner).
Mit 3.5 gewünschtem pH, 1 Pufferkapazität bei geduldeter Absenkung des pH um 0.3 (3.5 - 3.2 = 0.3) werden mit dem ersten Rechner 7g Citronensäure und 1g Natriumhydroxid berechnet.
Wenn man ein gutes pH-Messgerät hat, kann man diese Mischung nun noch überprüfen. Dies ist vor allem empfehlenswert, wenn die Pufferzutaten alst sind oder einen etwas feuchten Eindruck machen:
Angenommen man hat den Puffer auf 200ml Wasser angemischt, dann gibt man in den zweiten Rechner 0.2 liter Maische, 7g Citronensäure und 1g Natriumhydroxid ein und bekommt einen pH von 3.4 berechnet. Angenommen das pH-Messgerät zeigt in diesen 200ml nun 3.6 an, gibt man noch so viel Citronensäure hinzu, bis das Messgerät 3.4 anzeigt. Oder wenn man pH 3.2 misst, gibt man noch so viel Natriumhydroxid dazu, bis 3.4 angezeigt wird.
Der Puffer sollte in jedem Fall in etwas Wasser gelöst werden, bevor man ihn zur Maische gibt. Am besten löst man zuerst die Säure und gibt dann langsam die Base dazu.
Die Hefe sollte erst nach der Pufferung zur Maische gegeben werden.
Edit: Dieser Rechner hat mit der Zeit einige Veränderungen erfahren. So gibt es nun zwei verschiedene Pufferkapazitäten. Und die Anmerkung geht auch auf das von der Hefe produzierte CO2 ein.
Beziehungsweise sind es mehrere Rechner. Es geht um die Einstellung und Pufferung des pH von Maischen mit Hilfe von Zutaten wie Citronensäure und Natriumhydroxid.
Der Vorteil der Zugabe eines solchen Puffers verglichen mit der Zugabe zum Beispiel nur von Citronensäure oder auch Biogen M ist, daß er nicht nur den pH senkt, sondern auch einen Schutz gegen weiteres Absinken des pHs bietet.
Und der Vorteil gegenüber einer Zugabe zum Beispiel nur von Calciumcarbonat ist, daß man mit einem Puffer von Anfang an den pH gesenkt hat, nicht erst nachdem die Gärung in Schwung gekommen ist.
Alles weitere steht direkt in der Anmerkung des Rechners.
Hier aber noch ein praktisches Beispiel:
Angenommen, man möchte 10 liter Maische zwischen pH 3.5 und 3.2 vergären und den Puffer aus wasserfreie Citronensäure und Natriumydroxid herstellen:
Da man nicht erwartet, daß der pH ohne Puffer unter 3 sinken würde, reicht eine Pufferkapazität von 1 (siehe Anmerkung im Rechner).
Mit 3.5 gewünschtem pH, 1 Pufferkapazität bei geduldeter Absenkung des pH um 0.3 (3.5 - 3.2 = 0.3) werden mit dem ersten Rechner 7g Citronensäure und 1g Natriumhydroxid berechnet.
Wenn man ein gutes pH-Messgerät hat, kann man diese Mischung nun noch überprüfen. Dies ist vor allem empfehlenswert, wenn die Pufferzutaten alst sind oder einen etwas feuchten Eindruck machen:
Angenommen man hat den Puffer auf 200ml Wasser angemischt, dann gibt man in den zweiten Rechner 0.2 liter Maische, 7g Citronensäure und 1g Natriumhydroxid ein und bekommt einen pH von 3.4 berechnet. Angenommen das pH-Messgerät zeigt in diesen 200ml nun 3.6 an, gibt man noch so viel Citronensäure hinzu, bis das Messgerät 3.4 anzeigt. Oder wenn man pH 3.2 misst, gibt man noch so viel Natriumhydroxid dazu, bis 3.4 angezeigt wird.
Der Puffer sollte in jedem Fall in etwas Wasser gelöst werden, bevor man ihn zur Maische gibt. Am besten löst man zuerst die Säure und gibt dann langsam die Base dazu.
Die Hefe sollte erst nach der Pufferung zur Maische gegeben werden.
Edit: Dieser Rechner hat mit der Zeit einige Veränderungen erfahren. So gibt es nun zwei verschiedene Pufferkapazitäten. Und die Anmerkung geht auch auf das von der Hefe produzierte CO2 ein.
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Refluxspule
Ein neuer Rechner ist da: Refluxspulen
Er ist für die gedacht, welche mit dem Gedanken spielen, sich in der alten Kunst des Biegens von Refluxspulen zu versuchen, aber -wahrscheinlich zurecht- nicht einfach drauflosbiegen wollen.
In den Rechner gibt man ein, wie man sich die Spule vorstellt und dann berechnet er zusätzliche Informationen, wie die nötige Rohrlänge, die Anzahl der Windungen und anderes. Mit diesen Informationen kann man seinen Plan konkretisieren. Beim Biegen tauchen dann solche Situationen wie "Ich habe keine Ahnung, ob ich nicht schon längst den Übergang auf die äußere Spule hätte machen sollen." oder "Jetzt habe ich 50cm Rohr übrig. Hätte ich doch besser noch zwei Windungen mehr gemacht." erst gar nicht auf.
Er ist für die gedacht, welche mit dem Gedanken spielen, sich in der alten Kunst des Biegens von Refluxspulen zu versuchen, aber -wahrscheinlich zurecht- nicht einfach drauflosbiegen wollen.
In den Rechner gibt man ein, wie man sich die Spule vorstellt und dann berechnet er zusätzliche Informationen, wie die nötige Rohrlänge, die Anzahl der Windungen und anderes. Mit diesen Informationen kann man seinen Plan konkretisieren. Beim Biegen tauchen dann solche Situationen wie "Ich habe keine Ahnung, ob ich nicht schon längst den Übergang auf die äußere Spule hätte machen sollen." oder "Jetzt habe ich 50cm Rohr übrig. Hätte ich doch besser noch zwei Windungen mehr gemacht." erst gar nicht auf.
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Obstmaischerechner 2
Wir haben den Obstmaischerechner überarbeitet.
Bei sehr dickflüssigen Maischen ist Forumsmitgliedern aufgefallen, daß wesentlich höhere SG-Werte (oder Brix oder Oechsle usw.) gemessen als berechnet werden. Der Grund ist, daß unvergärbare Stoffe eine relativ hohe Dichte haben. Wir hatten diese Soffe zwar aus den Maischen rausgerechnet, aber nur, um den Alkoholgehalt und die mögliche Zuckerzugabe richtig einschätzen zu können. Nicht bei der Berechnung der Messwerte oder andersherum von den Messwerten zum Zuckergehalt, da wir fälschlicherweise dachten, daß ein Großteil der schweren unvergärbaren Stoffe vor dem Messen herausgefiltert werden kann.
Nun haben wir eine neue Berechnung, welche die unvergärbaren Stoffe komplett mitberücksichtigt und somit also Messwerte von ungefilterten Maischen berechnen kann bzw. von Messwerten an ungefilterten Maischen auf den Zuckergehalt schließen kann.
Dickflüssige Maischen sind schwer zu messen. Aber sie werden vor der Vergärung ja etwas verdünnt, was dann nicht nur die Gärung sondern nebenbei auch das Messen erleichtert. Deswegen kann man in den Rechner Messungen von bereits verdünnten Maischen angeben.
Da wir unser Obst vor der Gärung ja nicht analysieren lassen, kann die Berechnung natürlich nicht 100% genau sein. Je weiter wir uns von reinem Zuckerwasser entfernen, desto mehr Unabwägbarkeiten kommen hinzu, desto ungenauer ist die Rechnung. Also die Ungenauigkeiten dadurch, daß wir die Zusammensetzung unserer Früchte nicht genau kennen, sind bei einer Schlehenmaische höher als bei einer Apfelsaftmaische, da die Schlehen erstens Steine von uns nicht ganz genau bekannter Größe und Dichte enthalten und zweitens deren Fruchtfleisch verglichen mit Apfelsaft wesentlich mehr unvergärbare Stoffe enthält.
Obstmaischerechner
In der Anmerkung unter dem Rechner steht noch eine ganze Menge dazu. Es erklärt sich leider nicht von alleine, wenn man ihn zum ersten mal verwendet.
Unter der Anmerkung sind die hobbybrennen.ch sucht Daten von Steinobst! -Daten, welche im Forum gesammelt wurden, tabellarisch dargestellt. Auch der jeweilige Benutzername des Mitglieds, welches die Messung gemacht hat, steht da. Wem es nicht passt, daß sein Benutzername dort steht, kann mir das in einer PN schreiben. Auch ohne Begründung. Ich nehme den Namen dann heraus.
Weitere Entwicklungen können noch kommen:
- Die Dichte der unvergärbaren Stoffe genauer berechnen und vielleicht auch einen sortenspezifischen Wert zu ermitteln versuchen. Und dann rechnet der Rechner bei Quitten mit einer anderen Dichte der unvergärbaren Stoffe als bei Zwetschgen.
- Die Dichte der Steine ebenfalls sortenspezifisch machen. Dafür reichen die Messdaten aber noch nicht. Ich habe den Verdacht, Steine von unreifem Obst haben eine geringere Dichte als die von reifem Obst. Und Steine, die eng von Fruchtfleisch umschlossen sind wie zum Beispiel Schlehensteine, haben eine höhere Dichte als Steine, die eher locker in der Frucht liegen. Es gibt aber ein paar Messdaten, die dazu nicht passen.
Aber irgendwann stellt sich die Frage, ob solche Feinheiten verschwinden hinter den naturgegebenen Unterschieden der Früchte.
Bei sehr dickflüssigen Maischen ist Forumsmitgliedern aufgefallen, daß wesentlich höhere SG-Werte (oder Brix oder Oechsle usw.) gemessen als berechnet werden. Der Grund ist, daß unvergärbare Stoffe eine relativ hohe Dichte haben. Wir hatten diese Soffe zwar aus den Maischen rausgerechnet, aber nur, um den Alkoholgehalt und die mögliche Zuckerzugabe richtig einschätzen zu können. Nicht bei der Berechnung der Messwerte oder andersherum von den Messwerten zum Zuckergehalt, da wir fälschlicherweise dachten, daß ein Großteil der schweren unvergärbaren Stoffe vor dem Messen herausgefiltert werden kann.
Nun haben wir eine neue Berechnung, welche die unvergärbaren Stoffe komplett mitberücksichtigt und somit also Messwerte von ungefilterten Maischen berechnen kann bzw. von Messwerten an ungefilterten Maischen auf den Zuckergehalt schließen kann.
Dickflüssige Maischen sind schwer zu messen. Aber sie werden vor der Vergärung ja etwas verdünnt, was dann nicht nur die Gärung sondern nebenbei auch das Messen erleichtert. Deswegen kann man in den Rechner Messungen von bereits verdünnten Maischen angeben.
Da wir unser Obst vor der Gärung ja nicht analysieren lassen, kann die Berechnung natürlich nicht 100% genau sein. Je weiter wir uns von reinem Zuckerwasser entfernen, desto mehr Unabwägbarkeiten kommen hinzu, desto ungenauer ist die Rechnung. Also die Ungenauigkeiten dadurch, daß wir die Zusammensetzung unserer Früchte nicht genau kennen, sind bei einer Schlehenmaische höher als bei einer Apfelsaftmaische, da die Schlehen erstens Steine von uns nicht ganz genau bekannter Größe und Dichte enthalten und zweitens deren Fruchtfleisch verglichen mit Apfelsaft wesentlich mehr unvergärbare Stoffe enthält.
Obstmaischerechner
In der Anmerkung unter dem Rechner steht noch eine ganze Menge dazu. Es erklärt sich leider nicht von alleine, wenn man ihn zum ersten mal verwendet.
Unter der Anmerkung sind die hobbybrennen.ch sucht Daten von Steinobst! -Daten, welche im Forum gesammelt wurden, tabellarisch dargestellt. Auch der jeweilige Benutzername des Mitglieds, welches die Messung gemacht hat, steht da. Wem es nicht passt, daß sein Benutzername dort steht, kann mir das in einer PN schreiben. Auch ohne Begründung. Ich nehme den Namen dann heraus.
Weitere Entwicklungen können noch kommen:
- Die Dichte der unvergärbaren Stoffe genauer berechnen und vielleicht auch einen sortenspezifischen Wert zu ermitteln versuchen. Und dann rechnet der Rechner bei Quitten mit einer anderen Dichte der unvergärbaren Stoffe als bei Zwetschgen.
- Die Dichte der Steine ebenfalls sortenspezifisch machen. Dafür reichen die Messdaten aber noch nicht. Ich habe den Verdacht, Steine von unreifem Obst haben eine geringere Dichte als die von reifem Obst. Und Steine, die eng von Fruchtfleisch umschlossen sind wie zum Beispiel Schlehensteine, haben eine höhere Dichte als Steine, die eher locker in der Frucht liegen. Es gibt aber ein paar Messdaten, die dazu nicht passen.
Aber irgendwann stellt sich die Frage, ob solche Feinheiten verschwinden hinter den naturgegebenen Unterschieden der Früchte.
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Re: Rechner
Wir haben den Rechner ph-Puffer überarbeitet.
Es sind jetzt auch Malat-, Tartrat- und Ascorbatpuffer berechenbar. Also Puffer mit Apfel-, Wein- oder Ascorbinsäure (Vitamin C). Zumindest die Malatpuffer haben auch einen hohen praktischen Wert.
Als Basen können nur noch Natrium- und Kaliumverbindungen berechnet werden. Calciumverbindungen haben wir rausgenommen, weil sich diese mit dem bei der Gärung entstehenden CO2 zu Calciumcarbonat verbinden würden, welches dann fast vollständig ausfallen würde.
Näheres steht in der Anmerkung des Rechners.
Es sind jetzt auch Malat-, Tartrat- und Ascorbatpuffer berechenbar. Also Puffer mit Apfel-, Wein- oder Ascorbinsäure (Vitamin C). Zumindest die Malatpuffer haben auch einen hohen praktischen Wert.
Als Basen können nur noch Natrium- und Kaliumverbindungen berechnet werden. Calciumverbindungen haben wir rausgenommen, weil sich diese mit dem bei der Gärung entstehenden CO2 zu Calciumcarbonat verbinden würden, welches dann fast vollständig ausfallen würde.
Näheres steht in der Anmerkung des Rechners.
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Begleitstoffe-Flüchtigkeitsdiagramm
Ein neuer Rechner ist da: Begleitstoffe-Flüchtigkeitsdiagramm
Er stellt die Flüchtigkeit von Aromastoffen in Diagrammen dar, wie man sie auch hier und da zur Verdeutlichung des Verhaltens dieser Stoffe bei der Destillation in der Literatur finden kann.
Hier zum Beispiel eine Graphik aus The Alcohol Textbook:
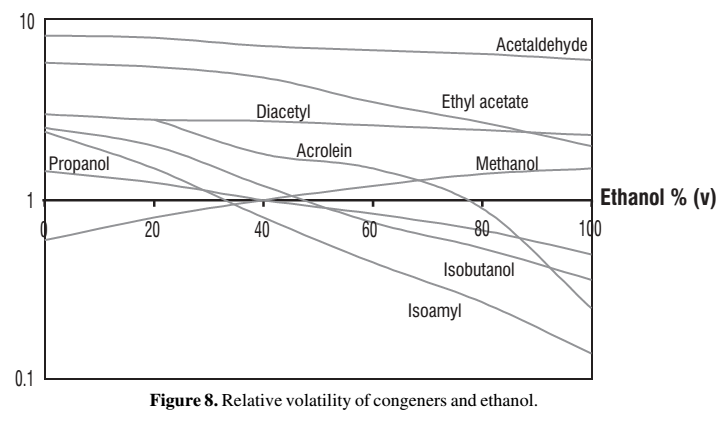
und hier aus Aus Whisky Technology, Production and Marketing:
Die x-Achse ist die Alkoholstärke im Kessel und die y-Achse die Flüchtigkeit. Im ersten Diagramm die Flüchtigkeit verglichen mit der Flüchtigkeit zu Ethanol. Im zweiten Diagramm die absolute Flüchtigkeit logarithmisch umgerechnet.
Solche Diagramme kann unser Rechner mit 49 Stoffen, Ethanol und Wasser darstellen. So zum Beispiel sieht es aus, wenn man ein Diagramm mit denselben Stoffen wie das erste oben dargestellt wird:
Daß die Kurven etwas anders verlaufen, als auf dem Bild oben, liegt daran, daß wir von vielen Stoffen mehrere Daten haben und diese Daten erstmal bewertet und dann kombiniert haben.
Außerdem kann man sich darstellen lassen, wie die Flüchtigkeiten auf Rektifikation reagieren.
Und man kann umschalten, daß auf der x-Achse nicht die vol% im Kessel sondern die vol% im Dampf sind.
Und man kann, wie bei unserem Begleitstoffesimulator 1, alle Stoffe miteinander vergleichen. So ist es zB sinnvoll sich alle Ester im Vergleich zum Ester Ethylacetat anzeigen zu lassen, da letzterer eher einen negativen Einfluß auf das Aroma hat und die anderen eher gute. Daraus kann man Schlüsse ziehen, welche Vor- und Nachteile die verscheidenen Destillationsprozesse haben. Viele Beispiele dazu und Informationen zu unseren Daten stehen in der Vorstellung des Begleitstofferechners ein paar Beiträge weiter oben.
Weitere Informationen gibt es auch noch in der Anmerkung unter dem Rechner.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
Er stellt die Flüchtigkeit von Aromastoffen in Diagrammen dar, wie man sie auch hier und da zur Verdeutlichung des Verhaltens dieser Stoffe bei der Destillation in der Literatur finden kann.
Hier zum Beispiel eine Graphik aus The Alcohol Textbook:
und hier aus Aus Whisky Technology, Production and Marketing:
Solche Diagramme kann unser Rechner mit 49 Stoffen, Ethanol und Wasser darstellen. So zum Beispiel sieht es aus, wenn man ein Diagramm mit denselben Stoffen wie das erste oben dargestellt wird:
Außerdem kann man sich darstellen lassen, wie die Flüchtigkeiten auf Rektifikation reagieren.
Und man kann umschalten, daß auf der x-Achse nicht die vol% im Kessel sondern die vol% im Dampf sind.
Und man kann, wie bei unserem Begleitstoffesimulator 1, alle Stoffe miteinander vergleichen. So ist es zB sinnvoll sich alle Ester im Vergleich zum Ester Ethylacetat anzeigen zu lassen, da letzterer eher einen negativen Einfluß auf das Aroma hat und die anderen eher gute. Daraus kann man Schlüsse ziehen, welche Vor- und Nachteile die verscheidenen Destillationsprozesse haben. Viele Beispiele dazu und Informationen zu unseren Daten stehen in der Vorstellung des Begleitstofferechners ein paar Beiträge weiter oben.
Weitere Informationen gibt es auch noch in der Anmerkung unter dem Rechner.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Re: Begleitstoffe-Flüchtigkeitstabellen
Wir beschäftigen uns nun schon lange immer wieder mit den Begleitstoffen. Das Ziel ist ein Rechner, mit dem man Mehrfachdestillationen mit Abtrennungen durchrechnen kann. Dieser Rechner ist im Prinzip auch schon fertig. Aber Ergebnisse der damit simulierten Mehrfachdestillationen haben immer wieder Fragen aufgeworfen und uns deswegen dazu gebracht, erstmal lieber wieder Schritte zurückzugehen und mit einfacheren Rechner lieber nochmal den Blick auf Details zu richten, anstelle das Gesamtbild anzuschauen. Diese Rechner werden trotz ihrer Einfachkeit vielleicht nicht oft hilfreich sein, da sie begrenzte Informationen liefern, aber wir veröffentlichen sie trotzdem, auch da wir selber nicht genau wissen, was nochmal wichtig wird.
Hier ist der erste:
Begleitstoffe-Flüchtigkeitstabellen
Das ist eine tabellarische Version des Rechners Begleitstoffe-Flüchtigkeitsdiagramm.
Ein interessantes Detail ist uns dabei für die Vorlaufabtrennung aufgefallen. Ein Hinweis, daß langsames Abtrennen des Vorlaufs mit passiver Rektifikation weniger hilft, als man vielleicht denkt:
Man stellt die vol% auf eine typische Feinbrandstärke wie 30vol% und die Rektifikation auf 0. Nun schaut man sich die wertvollen unteren Ester an und fährt dabie die Rektifikation langsam hoch. Man stellt fest, daß deren Flüchtigkeit relativ zu Ethanol entweder nur sehr langsam sinkt oder sogar langsam ansteigt und dann erst so ab Rektifikation 1 richtig stark abfällt. Das bedeutet, daß mit weniger als Rektifikation 1 -und mit passsiver Rektifikation ist normalerweise nicht mehr zu erreichen- ein großer Teil der wertvollen Ester im Vorlauf verlorengeht. Um das zu verhindern braucht man also aktive Rektifikation.
Diese Sache kann man auch mit unseren anderen Begleitstofferechnern herausbekommen. Hier aber ist sie sehr gut sichtbar.
Weitere Infos stehen in der Anmerkung direkt unter dem Rechner.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
Hier ist der erste:
Begleitstoffe-Flüchtigkeitstabellen
Das ist eine tabellarische Version des Rechners Begleitstoffe-Flüchtigkeitsdiagramm.
Ein interessantes Detail ist uns dabei für die Vorlaufabtrennung aufgefallen. Ein Hinweis, daß langsames Abtrennen des Vorlaufs mit passiver Rektifikation weniger hilft, als man vielleicht denkt:
Man stellt die vol% auf eine typische Feinbrandstärke wie 30vol% und die Rektifikation auf 0. Nun schaut man sich die wertvollen unteren Ester an und fährt dabie die Rektifikation langsam hoch. Man stellt fest, daß deren Flüchtigkeit relativ zu Ethanol entweder nur sehr langsam sinkt oder sogar langsam ansteigt und dann erst so ab Rektifikation 1 richtig stark abfällt. Das bedeutet, daß mit weniger als Rektifikation 1 -und mit passsiver Rektifikation ist normalerweise nicht mehr zu erreichen- ein großer Teil der wertvollen Ester im Vorlauf verlorengeht. Um das zu verhindern braucht man also aktive Rektifikation.
Diese Sache kann man auch mit unseren anderen Begleitstofferechnern herausbekommen. Hier aber ist sie sehr gut sichtbar.
Weitere Infos stehen in der Anmerkung direkt unter dem Rechner.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Begleitstoffe-Fraktionssimulator
Und hier ist der zweite:
Begleitstoffe-Fraktionssimulator
Dieser rechnet zuerst mit der Vorgabe, wie viel liter und vol% man in der Destille hat und wie stark die Destille rektifiziert, eine komplette Destillation durch. Danach kann man mit einem Schieberegler einen Abtrennpunkt festlegen bzw. verschieben und in den Tabellen beobachten, wie sich die Stoffe auf den Abschnitt vor oder nach dem Schnitt verteilen.
Der Sinn kann zum Beispiel sein, sich darstellen zu lassen, was es ausmacht, ob man mehr oder weniger Vorlauf abtrennt. Die Ergebnisse bleiben meiner Meinung nach aber recht abstrakt. Aber einige, zum Teil aber vielleicht triviale Sachen, kann man sehr gut erkennen:
Wenn man sich die Werte vor dem Schieberegler anschaut und diesen erstmal ganz links hat und dann einen Schritt nach rechts fährt, dann sieht man die Zusammensetzung des allerersten Tropfen Destillats. In den Feldern mit relativen Prozentzahlen gibt es nun welche, wo unter 100%, und welche, wo über 100% drinnensteht. Wenn man nun mit dem Schieberegler nach rechts fährt, also eine immer größer werdende Menge Destillat aufnimmt, wandern alle diese Zahlen in Richtung 100. Spätestens am Ende (bei leergebranntem Kessel) sind alle bei 100%. Daß am Ende alle bei 100% landen, kann natürlich auch nicht anders sein. Aber:
Es gibt keinen einzigen Fall, wo sich die die Richtung mal ändert, also wo sich ein Wert von 100 wegbewegt, oder wo ein Wert mal egal von unten oder von oben über die 100 hinweggeht und dann später wieder umdreht.
Das bedeutet folgendes:
1. Je kürzer eine Fraktion ist, desto individueller ist sie. Denn dann sind die relativen %-Werte weit von 100% entfernt. Individuell meint, anders als der Kesselinhalt.
2. Gute Eigenschaften von Fraktionen werden weniger gut und schlechte Eigenschaften weniger schlecht, wenn man diese Fraktionen verlängert.
3. Eine kurze, sehr individuelle Fraktion zu verwerfen (zB beim Vorlauf), macht aber bei dem verbleibenden Rest trotzdem weniger aus, als wenn man diese Fraktion verlängert, sie also uninteressanter werden lässt. Die Menge sticht. Während sich die relativen Werte links vom Schieberegler an die 100% annähern, entfernen sich die relativen Werte rechts vom Regler immer weiter von den 100%, der Rest wird also individueller.
4. Wenn man sich zB einen Vorlauf darstellen lässt, dann liefert der erste Wert am Schieberegler schon fast die gesamte Information, wie dieser Vorlauf charakterlich sein wird, und damit, wie gut die Situation (also die eingegebenen vol% im Kessel und die Rektifikation) zum Vorlaufabtrennen geeignet ist. Natürlich ist dann auch die Menge wichtig, die man abtrennt. Und die Situation hat auch Einfluß auf die nötige Menge, vor allem die Rektifikation. Aber grundsätzlich kann eine ungünstige Situation niemals durch die Menge in eine günstige verwandelt werden. An der Menge zu drehen, wäre natürlich einfach, die Situation kann man aber nur durch vorausschauendes Planen beeinflussen.
Es kann sinnvoll sein, mit diesem Rechner mit mehreren Registerkarten verschiedene Szenarien vergleichend durchzurechnen. So kann man zum Beispiel schauen, was für Auswirkungen die Rektifikation und die Alkoholstärke auf die Vorlaufabtrennung hat. Hier eine Ergebnistabelle dazu:
Vier verscheidene Alkoholstärken und drei verschiedene Rektifikationsstufen. Abgetrennt wurde, sobald die Summe des Acetaldehyds und des Ethylacetats (also der beiden wichtigsten Vorlaufstoffen) 180% ergeben. Angezeigt werden, wie viel% des Alkohols man im Vorlauf verliert ("%Eth") und wieviel % der drei wetvollen Ester Ethylcaproat, Ethylcaprylat und Ethylcaprinat ("%C") im Schnitt (also die Summe der drei Werte durch drei geteilt), welche möglichst nicht im Vorlauf landen sollen.
Man sieht, daß man mit möglichst hoher Rektifikation die besten Ergebnisse bekommt (am wenigsten %Eth und am wenigsten %C im Vorlauf ist). Das ist keine Überraschung. Überraschend ist aber, daß hohe vol% im Kessel zur Folge haben, daß man mehr Alkohol verliert, um die geforderte Menge Acetaldehyd und Ethylacetat abzutrennen. Wenn man sich die Werte im Simulator genauer anschaut, merkt man, daß das Problem das Ethylacetat ist. Dieses lässt sich generell sehr schlecht bei hohen Prozent im Kessel abtrennen. Also das Acetaldehyd lässt sich zwar sehr gut abtrennen bei hohen vol%, das Ethylacetat aber nicht. Allgemein lassen sich die Ester schlecht bei hohen vol% abtrennen. Deswegen verliert man auch von den guten Estern mehr, wenn die vol% im Kessel niedrig sind. Bei hoher Rektifikation macht das aber nicht mehr viel aus.
Die Ergebnisse, welche wir schon mit dem Begleitstoffesimulator 1 veröffentlicht haben, könnte man mit diesem Rechner auch gut nachstellen. Aber das wäre eine Wiederholung.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
Begleitstoffe-Fraktionssimulator
Dieser rechnet zuerst mit der Vorgabe, wie viel liter und vol% man in der Destille hat und wie stark die Destille rektifiziert, eine komplette Destillation durch. Danach kann man mit einem Schieberegler einen Abtrennpunkt festlegen bzw. verschieben und in den Tabellen beobachten, wie sich die Stoffe auf den Abschnitt vor oder nach dem Schnitt verteilen.
Der Sinn kann zum Beispiel sein, sich darstellen zu lassen, was es ausmacht, ob man mehr oder weniger Vorlauf abtrennt. Die Ergebnisse bleiben meiner Meinung nach aber recht abstrakt. Aber einige, zum Teil aber vielleicht triviale Sachen, kann man sehr gut erkennen:
Wenn man sich die Werte vor dem Schieberegler anschaut und diesen erstmal ganz links hat und dann einen Schritt nach rechts fährt, dann sieht man die Zusammensetzung des allerersten Tropfen Destillats. In den Feldern mit relativen Prozentzahlen gibt es nun welche, wo unter 100%, und welche, wo über 100% drinnensteht. Wenn man nun mit dem Schieberegler nach rechts fährt, also eine immer größer werdende Menge Destillat aufnimmt, wandern alle diese Zahlen in Richtung 100. Spätestens am Ende (bei leergebranntem Kessel) sind alle bei 100%. Daß am Ende alle bei 100% landen, kann natürlich auch nicht anders sein. Aber:
Es gibt keinen einzigen Fall, wo sich die die Richtung mal ändert, also wo sich ein Wert von 100 wegbewegt, oder wo ein Wert mal egal von unten oder von oben über die 100 hinweggeht und dann später wieder umdreht.
Das bedeutet folgendes:
1. Je kürzer eine Fraktion ist, desto individueller ist sie. Denn dann sind die relativen %-Werte weit von 100% entfernt. Individuell meint, anders als der Kesselinhalt.
2. Gute Eigenschaften von Fraktionen werden weniger gut und schlechte Eigenschaften weniger schlecht, wenn man diese Fraktionen verlängert.
3. Eine kurze, sehr individuelle Fraktion zu verwerfen (zB beim Vorlauf), macht aber bei dem verbleibenden Rest trotzdem weniger aus, als wenn man diese Fraktion verlängert, sie also uninteressanter werden lässt. Die Menge sticht. Während sich die relativen Werte links vom Schieberegler an die 100% annähern, entfernen sich die relativen Werte rechts vom Regler immer weiter von den 100%, der Rest wird also individueller.
4. Wenn man sich zB einen Vorlauf darstellen lässt, dann liefert der erste Wert am Schieberegler schon fast die gesamte Information, wie dieser Vorlauf charakterlich sein wird, und damit, wie gut die Situation (also die eingegebenen vol% im Kessel und die Rektifikation) zum Vorlaufabtrennen geeignet ist. Natürlich ist dann auch die Menge wichtig, die man abtrennt. Und die Situation hat auch Einfluß auf die nötige Menge, vor allem die Rektifikation. Aber grundsätzlich kann eine ungünstige Situation niemals durch die Menge in eine günstige verwandelt werden. An der Menge zu drehen, wäre natürlich einfach, die Situation kann man aber nur durch vorausschauendes Planen beeinflussen.
Es kann sinnvoll sein, mit diesem Rechner mit mehreren Registerkarten verschiedene Szenarien vergleichend durchzurechnen. So kann man zum Beispiel schauen, was für Auswirkungen die Rektifikation und die Alkoholstärke auf die Vorlaufabtrennung hat. Hier eine Ergebnistabelle dazu:
| Kessel | Rektifikation = 0 | Rektifikation = 1 | Rektifikation = 3 |
| 60vol% | 37.5%Eth 34.2%C | 13.8%Eth 4.5%C | 2.2%Eth 0.0%C |
| 35vol% | 31.3%Eth 93.9%C | 12.2%Eth 18.7%C | 1.3%Eth 0.1%C |
| 15vol% | 32.7%Eth 98.0%C | 7.8%Eth 61.5%C | 0.7%Eth 0.4%C |
| 5vol% | 36.1%Eth 98.5%C | 7.1%Eth 97.0%C | 0.5%Eth 1.6%C |
Man sieht, daß man mit möglichst hoher Rektifikation die besten Ergebnisse bekommt (am wenigsten %Eth und am wenigsten %C im Vorlauf ist). Das ist keine Überraschung. Überraschend ist aber, daß hohe vol% im Kessel zur Folge haben, daß man mehr Alkohol verliert, um die geforderte Menge Acetaldehyd und Ethylacetat abzutrennen. Wenn man sich die Werte im Simulator genauer anschaut, merkt man, daß das Problem das Ethylacetat ist. Dieses lässt sich generell sehr schlecht bei hohen Prozent im Kessel abtrennen. Also das Acetaldehyd lässt sich zwar sehr gut abtrennen bei hohen vol%, das Ethylacetat aber nicht. Allgemein lassen sich die Ester schlecht bei hohen vol% abtrennen. Deswegen verliert man auch von den guten Estern mehr, wenn die vol% im Kessel niedrig sind. Bei hoher Rektifikation macht das aber nicht mehr viel aus.
Die Ergebnisse, welche wir schon mit dem Begleitstoffesimulator 1 veröffentlicht haben, könnte man mit diesem Rechner auch gut nachstellen. Aber das wäre eine Wiederholung.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Begleitstoffesimulator 2
Nun ist endlich unser Begleitstoffesimulator 2 fertig!
Ein sehr interessanter Rechner, mit dem man bisher ungeklärte Fragen beantworten kann, bisher Unbewiesenes begründen kann und auch manches Neue entdecken kann.
Der Rechner kann Mehrfachdestillationen mit Abtrennungen und auch Wasserzugaben zwischen den Bränden durchrechnen und somit den Einfluss verschiedener Brennweisen auf Aromastoffe hin untersuchen.
Wer den Destillationssimulator 1 kennt, wird auch den neuen Rechner schnell verstehen, denn er baut auf ihm auf und ist in der Optik und Bedienung daher ähnlich.
In der Anmerkung unter dem Rechner stehen weitere Erklärungen zur Benutzung.
Rechenbeispiele und Ergebnisse:
10l mit 10vol%.
A)
1.Destillation bis 3.5l 0.1R.
2.Destillation bis 0.03l 0.3R, dann bis akt. 60vol% mit 0.1R, Vorlauf weg:
B)
1.Destillation bis 0.01l 0.5R, dann bis 3.49l 0.1R, Vorlauf weg.
2.Destillation bis 0.02l 0.4R, dann bis akt. 60vol% mit 0.1R, Vorlauf weg:
B ist allgemein etwas neutraler. Weniger Begleitstoffe verglichen mit Ethanol.
B ist qualitativ etwas schlechter, minimal bei den unteren Aldehyden (Valeraldehyd, Isovaleraldehyd) verglichen mit Acetaldehyd, und bei fast allen Estern im Vergleich zu Ethylacetat, und bei einigen Terpenen (Sonstige) macht es auch was aus. Der sehr kleine Unterschied des Destillationsprozesses macht aber natürlich nicht viel aus. Aber generell sieht man schon, daß ein Abtrennen des Vorlaufs teilweise beim Raubrand erstens einen etwas sauberen Brand erzeugt, zweitens aber eher die guten als die schlechten Aromastoffe abgetrennt werden.
Wenn man also mit einer Potstill Neutralalkohol brennt, sollte man jeden Brand für die Vorlaufabtrennung nutzen, denn die unterschiedliche Alkoholstärke jedesmal hilft; manche Stoffe lassen sich eher bei niedriger Alkoholstärke gut abtrennen und manche bei eher hoher Alkoholstärke.
Bei Aromabränden mit einer Potstill sollte man nur den letzten Brand, also den mit der höchsten Alkoholstärke, zum Vorlaufabtrennen nehmen, da nur eine hohe Alkoholstärke bewirkt, daß mehr schlechte als gute Stoffe im Vorlauf landen.
10l mit 30vol%.
Da der Vorlauf rektifiziert natürlich viel konzentrierter ist, muss ich bei der Destillation mit Rektifikation wenig abtrennen und bei dem ohne viel, damit das Ergebnis am Ende vergleichbar ist:
A)
0.2l 0.3R, dann bis akt. 60vol% 0.1R, Vorlauf weg:
B)
0.02l 5R, dann bis akt. 60vol% 0.1R, Vorlauf weg:
Mit Rektifikation sind die kleineren, schlechten Aldehyde fast vollkommen weg. Von den wertvollen sind aber noch etwa dieselben Mengen da, wie bei ohne Rektifikation.
Mit Rektifikation ist Ethylacetat fast vollkommen weg, ohne nur die Hälfte. Weiter unten die großen, guten Ester sind mit Rektifikation zum Teil komplett im Mittellauf gelandet, ohne Rektifikation nur im Schnitt so zu 60%.
Kaum Unterschiede bei den Säuren und Alkoholen.
Mit Rektifikation ist Aceton weg und leider auch a-Pinene. Diacetyl ist halbiert.
Mit Rektifikation 74 anstelle 69% Ethanolausbeute.
Also fast nur gutes bringt die Rektifikation. Ganz stark bei den Estern.
Das bedeutet auch, daß mit der Rektifikation des Vorlaufs Probleme gelöst werden können, welche bei Brennweisen, die bei anderen Aspekten, wie zum Beispiel der Menge an Säuren, Vorteile bieten, aber bei den Estern und Aldehyden nicht optimal sind. Also wenn man die Möglichkeit hat, den Vorlauf zu rektifizieren, kann man sich bei der Brennweise auf andere Dinge konzentrieren.
10l mit 10vol%.
Um ein deutliches Ergebnis zu bekommen, wird nur wenig Vorlauf abgetrennt und recht spät der Mittellauf beendet.
A)
Raubrand: bis 3l 0.1R.
Feinbrand: bis 0.03l 5R, bis 90% des Alkohols destilliert ist 5R, Vorlauf weg:
B)
Raubrand: bis 3l 0.1R, dann 3l Wasser dazu.
Feinbrand: bis 0.03l 5R, bis 90% des Alkohols destilliert ist 5R, Vorlauf weg:
Mit Wasser sind die Aldehyde etwas besser abgetrennt.
Mit Wasser sind wesentlich weniger niedrige Ester übrig, aber teilweise mehr von den besseren.
Ohne Wasser sind die höheren Alkohole etwas besser unterdrückt.
Bei den Sonstigen fällt D-Limonen auf, welches durch die Verdünnung stark in den Vorlauf gedrängt wurde, also weg ist.
Fazit: Verdünnen ist vom Geruch her neutraler und besser. Das Plus an höheren Alkoholen wird sich aber vielleicht negativ bemerkbar machen.
10l mit 19vol%.
A)
Einfachbrand bis 0.05l 0.3R, dann bis 91°C 0.1R, Vorlauf weg:
B)
Raubrand bis 3.5l 0.1R.
Feinbrand bis 0.04l 0.3R, dann bis akt. 70vol% 0.1R, Vorlauf weg:
Der Doppelbrand hat 77 anstelle 49% Ethanolausbeute.
Der Doppelbrand hat etwas mehr Aldehde, vor allem aber von den guten. Im Vergleich zu Ethanol sind es allerdings etwas weniger.
Bei den Estern das gleiche.
Der Doppelbrand hat viel weniger Säuren, ist also viel weicher auf der Zunge.
Der Doppelbrand hat mehr höhere Alkohole, im Vergleich zu Ethanol aber weniger.
Fazit: Durch die sehr unterschiedliche Ethanolausbeute sind die beiden Brände sehr schlecht vergleichbar. Dadurch, daß man beim Einfachbrand so viel Ethanol verwirft, verteilen sich die flüchtigen Aromen auf weniger Alkohol. Das ist ein Vorteil vom Einfachbrand. Allerdings ist sein Säuregehalt sehr hoch, was ihn kratzig macht. Außerdem ist das Verhältnis von guten zu schlechten Stoffen innerhalb einer Stoffgruppe beim Doppelbrand meist besser.
Wenn man, um die Ausbeute beim Einfachbrand zu erhöhen, den den Mittellauf später beendet, verliert man den Vorteil des Einfachbrands, daß sich die Aromen auf weniger Alkohol verteilen, und es kommen immer mehr Säuren. Die niedrige Ausbeute rettet also den Einfachbrand. Ohne das Opfern dieses Ethanols verliert man Qualität.
Nun könnte man beim Doppelbrand meinen, daß man beim Feinbrand nicht so hohe vol% im Kessel haben sollte. Dafür muss man entweder zwischen den Bränden verdünnen oder den Raubrand verlängern.
C)
Doppelbrand wie B, aber zwischen den Bränden 1l Wasser dazu und beim Feinbrand wegen des niedrigeren Alkoholgehalts im Kessel erst bei 65vol% aktuellem Alkoholgehalt abgetrennt:
Das ist bei den Aldehyden und Alkoholen etwas schlechter und bei den Estern etwas besser. Und hat eine etwas geringere Ethanolausbeute. Bei den Säuren schaut es schlechter aus, aber bei der wichtigsten Säure, der Essigsäure, ist es sogar etwas besser.
D)
Doppelbrand wie C, aber anstelle 1l Wasser dazuzugeben wird 1l mehr raugebrannt:
Das bringt vor allem viel mehr Säuren. Und etwas mehr Ethanol und etwas mehr höhere Alkohole. Schaut nicht gut aus.
Gesamtfazit: Der Einfachbrand schaut bei den Säuren und bei der Mittellaufausbeute sehr schlecht aus. Aber die schlechte Ethanolausbeute rettet ihn in gewisser Weise. Erhöht man diese, wird er sauer. Beim Doppelbrand scheint das extrem weit herunterbrennen, um nicht allzu hohe vol% im Kessel zu haben, eine schlechte Empfehlung zu sein. Weniger weit herunterzubrennen und dann entweder verdünnt oder unverdünnt zu brennen, scheint beides besser zu sein.
10l Maische mit 8vol%.
A)
Raubrand bis 2.5l 0.1R, also ein sehr kurzer Raubrand. Dann beim Feinbrand 0.03l mit 0.4R Vorlauf, Mittellauf bis 61vol% aktueller Alkoholgehalt:
B)
Raubrand bis 3.5l 0.1R, also ein langer Raubrand. Dann beim Feinbrand 0.03l mit 0.4R Vorlauf, Mittellauf bis 58vol% aktueller Alkoholgehalt. 58 anstelle 61vol% Mittellaufende, da der Alkoholgehalt des Raubrands niedriger ist:
Der kurze Raubrand hat weniger schlechte und mehr gute Aldehyde.
Und mehr von den guten Estern.
Etwas weniger Alkohole und deutlich weniger Säuren.
Er ist also sauberer trotz höherer Alkoholausbeute.
Es schaut also so aus, als bekommt man beim Doppelbrand mit langem Raubrennen mehr Geschmack, aber tendenziell etwas mehr schlechte Geschmäcker als gute.
Interessant ist vielleicht noch Phenol:
Dies ist bei dem langen Raubrand deutlich mehr vorhanden, 30.4 anstelle 22.9% im Vergleich zu Ethanol. Bei einer rauchigen Malzmaische bewirkt ein langer Raubrand also einen höheren Rauchgehalt im Whisky.
Zuerst sehr niedrigem Alkoholgehalt, 10l Maische mit 5vol%:
A)
Doppelbrand. Raubrand bis 2l mit 0.1R, dann Feinbrand 0.03l mit 0.4R Vorlauf, Mittellauf mit 0.1R bis 58vol% aktuellem Alkoholgehalt:
B)
Dreifachbrand. Raubrand bis 3l mit 0.1R, dann Mittelbrand bis 1.5l mit 0.1R, dann Feinbrand 0.03l mit 0.4R Vorlauf, Mittellauf mit 0.1R bis 65vol% aktuellem Alkoholgehalt:
Die Ethanolausbeute ist beim Dreifachbrand etwas höher, 65 anstelle 59%.
Es wurden etwas mehr schlechte Aldehyde mit dem Vorlauf abgetrennt, von den guten wurde weniger abgetrennt.
Ester wurden insgesamt weniger abgetrennt. Das betrifft aber vor allem die guten Ester.
Von den kleinen Säuren wurde viel mehr abgetrennt. Von den großen nur etwas weniger.
Es wurden im Vergleich zu Ethanol etwas mehr höhere Alkohole abgetrennt, allerdings etwas weniger Methanol.
Bei den Sonstigen Stoffen ist es unterschiedlich. Das Dreifachbrennen reduziert nicht zwingend diese Aromen. Zum Beispiel auch mit den Tails in Verbindung gebrachte Aromen wie Phenol werden nicht reduziert.
Das ist also ein klarer Sieg für den Dreifachbrand.
Ist das aber bei höherprozentiger Maische auch so?
10l Maische mit 12vol%:
C)
Doppelbrand. Raubrand bis 3.3l mit 0.1R, dann Feinbrand 0.05l mit 0.4R Vorlauf, Mittellauf mit 0.1R bis 66vol% aktuellem Alkoholgehalt:
D)
Dreifachbrand. Raubrand bis 4l mit 0.1R, dann Mittelbrand bis 2.5l mit 0.1R, dann Feinbrand 0.05l mit 0.4R Vorlauf, Mittellauf mit 0.1R bis 71vol% aktuellem Alkoholgehalt:
Die Ergebnisse sind ähnlich wie bei niedrigem Alkoholgehalt. Aber bei den Estern ist der Vorteil des Dreifachbrands verschwunden. Bei den Säuren ist der Unterschied vielleicht noch etwas größer geworden. Die Sonstigen wurden beim Dreifachbrand nun tendenziell mehr abgetrennt, was wahrscheinlich eher schlecht ist.
Das ist also kein so klarer Sieg mehr für den Dreifachbrand wie bei niedrigen vol%, aber immer noch leicht ersichtlich.
Ob der Doppelbrand irgendwann im Vorteil ist, wenn man die vol% in der Maische weiter erhöht?
10l Maische mit 19vol%:
E)
Doppelbrand. Raubrand bis 3.5l mit 0.1R, dann Feinbrand 0.05l mit 0.4R Vorlauf, Mittellauf mit 0.1R bis 72vol% aktuellem Alkoholgehalt:
F)
Dreifachbrand. Raubrand bis 4l mit 0.1R, dann Mittelbrand bis 3l mit 0.1R, dann Feinbrand 0.06l mit 0.4R Vorlauf, Mittellauf mit 0.1R bis 76vol% aktuellem Alkoholgehalt:
Der Dreifachbrand hat nur noch einen minimalen Vorteil bei den Aldehyden, bei den Estern ist er qualitativ schlechter, bei den Säuren immer noch viel besser, auch bei den Alkoholen. Die Sonstigen werden teilweise sehr stark unterdrückt. Er wird weicher als der Doppelbrand sein, aber vom Geruch schlechter.
Fazit: Es schaut so aus, als wäre der Dreifachbrand meistens besser. Allerdings schmilzt sein Vorteil, je alkoholstärker die Maische ist. Bei sehr niedrigprozentiger Maische ist er klar im Vorteil, bei Turbomaische ist es wohl Geschmackssache.
Dieser Vergleich ist schwer, fair durchzuführen. Denn wenn man einen Einfachbrand mit einer Refluxdestille macht, wird man den Vorlauf wahrscheinlich mit viel Reflux abtrennen. Diese Option hat man mit einer normalen Potstill aber nicht. Für einen Vergleich aber ist es interessant, wenn die Refluxdestille testweise zuerst mal auf diese Möglichkeit verzichtet:
10l mit 10vol%.
A)
Potstill-Doppelbrand, Raubrand 3l mit 0.1R, Feinbrand Vorlauf 0.05l mit 0.4R und dann bis 65vol% aktuellem Alkoholgehalt mit 0.1R:
B)
Reflux Einfachbrand, Feinbrand Vorlauf 0.05l mit 0.8R und dann versucht den Alkoholgehalt nicht unter 70vol% sinken zu lassen, bis man 64% des Alkohols als Mittellauf hat. Das ist das Protokoll:
Und das das Endergebnis:
Der Doppelbrand hat wesentlich mehr Aldehyde, auch von den schlechten, aber vor allem von den guten.
Er hat wesentlich mehr Ester, ebenfalls auch von den schlechten, aber vor allem von den guten.
Er hat weniger Essigsäure, aber mehr von den größeren Säuren. Da Essigsäure den Löwenanteil ausmacht, ist er etwas weicher.
Bei den Alkoholen sind beide Brennweisen ähnlich, der Doppelbrand hat etwas mehr Methanol.
Bei den Sonstigen gibt es ein paar, wo der Doppelbrand deutlich besser abschneidet.
Der Doppelbrand hat also qualitative Vorzüge, braucht aber wahrscheinlich eine etwas großzügigere Vorlaufabtrennung, um mehr von den kleinen Aldehyden und Estern abzutrennen.
Aber dieser Vergleich war ja unfair, weil die Refluxdestille auf ihre Möglichkeit, den Vorlauf zu rektifizieren, verzichtet hat.
Deswegen nun
C)
das gleiche Einfachbrand-Protokoll aber mit 0.02l Vorlauf mit 3R:
Die Probleme mit den Aldehyden und den Estern sind nun gelöst. Auch das Aceton ist weg. Das Problem mit der Essigsäure bleibt aber.
Ein Nachteil ist die nun etwas erhöhte Menge an höheren Alkoholen.
Der Reflux-Einfachbrand mit viel Reflux beim Vorlauf ist also besser in der Nase, der Potstill-Doppelbrand aber ist weicher.
Wenn man nun beim Doppelbrand ebenfalls den Vorlauf rektifizieren kann, bekommt man best of both worlds:
D)
Wie A, aber Vorlauf 0.02l mit 3R:
Es sind sehr viele der großen Aldehyde übrig, obwohl die kleinen fast weg sind. Ethylacetat wurde weniger gut abgetrennt als bei C, aber die größeren Ester wurden mehr geschont. Vielleicht hätte man 0.03 anstelle 0.02l Vorlauf eingeben sollen. Auch hier ist der Nachteil, daß die höheren Alkohole im Mittellauf etwas ansteigen, wenn man den Vorlauf rektifiziert. Die Vorteile bei den Säuren bleiben. Der Doppelbrand mit Rektifizierung des Vorlaufs ist also ähnlich gut oder vielleicht sogar besser in der Nase als der Reflux-Einfachbrand mit Rektifizierung des Vorlaufs und er ist weicher als der Reflux-Einfachbrand.
10l mit 8vol%.
A)
Raubrand bis 3l 0.1R, Mittelbrand bis 1.5l 0.1R:
Ergibt 1.5l mit 52.7vol%.
B)
Reflux-Raubrand bis 1.5l 52.7vol%. Dafür sind 0.63R nötig:
Bei beiden Versionen sind so gut wie alle Aldehyde, Ester, Alkohole und Sonstige im Destillat. Beim Doppelbrand ein bisschen vollständiger, was tendenziell etwas besser ausschaut. Aber bei den Säuren gibt es große Unterschiede. Der Doppelbrand unterdrückt die Essigsäure und Ameisensäure besser, der Einfachbrand eher die größeren Säuren. Da die Essigsäure am Häufigsten ist, wird der Doppelbrand etwas weicher sein. Außerdem hat er mehr von den interessanten Säuren mit höherem Potential, welche beim Feinbrand dann vielleicht wertvolle Ester bilden.
Nun könnte man den Refluxbrand aber auch mit ansteigendem Reflux machen, die Alkoholstärke also nie unter einen bestimmten Punkt fallen lassen. Hier das Protokoll:
C)
40vol% sollen also nicht unterschritten werden, am Ende sogar 45vol%. Wegen der Vergleichbarkeit soll die gleiche Alkoholstärke und Destillatmenge wie bei A und B erreicht werden.
Das Endergebnis:
Die Ergebnisse von A, B und C bilden Linien. Bei Essigsäure ist A zum Beispiel bei 3.7%, B bei 4.4% und C nun bei 4.7%. Oder Phenol ist bei A 67.8%, bei B 55.9% und bei C nun 46.4%.
Das ist für mich überraschend. Ich dachte, C würde bezüglich der Säuren sauberer sein. Hinsichtlich der wichtigen Essigsäure ist aber das Gegenteil passiert.
Wenn man B also schlechter als A findet (was wahrscheinlich ist), wird man C noch schlechter finden.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
Ein sehr interessanter Rechner, mit dem man bisher ungeklärte Fragen beantworten kann, bisher Unbewiesenes begründen kann und auch manches Neue entdecken kann.
Der Rechner kann Mehrfachdestillationen mit Abtrennungen und auch Wasserzugaben zwischen den Bränden durchrechnen und somit den Einfluss verschiedener Brennweisen auf Aromastoffe hin untersuchen.
Wer den Destillationssimulator 1 kennt, wird auch den neuen Rechner schnell verstehen, denn er baut auf ihm auf und ist in der Optik und Bedienung daher ähnlich.
In der Anmerkung unter dem Rechner stehen weitere Erklärungen zur Benutzung.
Rechenbeispiele und Ergebnisse:
Vergleich Doppelbrand, Vorlauf teilweise beim Raubrand abtrennen oder nicht?
10l mit 10vol%.
A)
1.Destillation bis 3.5l 0.1R.
2.Destillation bis 0.03l 0.3R, dann bis akt. 60vol% mit 0.1R, Vorlauf weg:
B)
1.Destillation bis 0.01l 0.5R, dann bis 3.49l 0.1R, Vorlauf weg.
2.Destillation bis 0.02l 0.4R, dann bis akt. 60vol% mit 0.1R, Vorlauf weg:
B ist allgemein etwas neutraler. Weniger Begleitstoffe verglichen mit Ethanol.
B ist qualitativ etwas schlechter, minimal bei den unteren Aldehyden (Valeraldehyd, Isovaleraldehyd) verglichen mit Acetaldehyd, und bei fast allen Estern im Vergleich zu Ethylacetat, und bei einigen Terpenen (Sonstige) macht es auch was aus. Der sehr kleine Unterschied des Destillationsprozesses macht aber natürlich nicht viel aus. Aber generell sieht man schon, daß ein Abtrennen des Vorlaufs teilweise beim Raubrand erstens einen etwas sauberen Brand erzeugt, zweitens aber eher die guten als die schlechten Aromastoffe abgetrennt werden.
Wenn man also mit einer Potstill Neutralalkohol brennt, sollte man jeden Brand für die Vorlaufabtrennung nutzen, denn die unterschiedliche Alkoholstärke jedesmal hilft; manche Stoffe lassen sich eher bei niedriger Alkoholstärke gut abtrennen und manche bei eher hoher Alkoholstärke.
Bei Aromabränden mit einer Potstill sollte man nur den letzten Brand, also den mit der höchsten Alkoholstärke, zum Vorlaufabtrennen nehmen, da nur eine hohe Alkoholstärke bewirkt, daß mehr schlechte als gute Stoffe im Vorlauf landen.
Vergleich Feinbrand, Vorlauf mit oder ohne Rektifikation
10l mit 30vol%.
Da der Vorlauf rektifiziert natürlich viel konzentrierter ist, muss ich bei der Destillation mit Rektifikation wenig abtrennen und bei dem ohne viel, damit das Ergebnis am Ende vergleichbar ist:
A)
0.2l 0.3R, dann bis akt. 60vol% 0.1R, Vorlauf weg:
B)
0.02l 5R, dann bis akt. 60vol% 0.1R, Vorlauf weg:
Mit Rektifikation sind die kleineren, schlechten Aldehyde fast vollkommen weg. Von den wertvollen sind aber noch etwa dieselben Mengen da, wie bei ohne Rektifikation.
Mit Rektifikation ist Ethylacetat fast vollkommen weg, ohne nur die Hälfte. Weiter unten die großen, guten Ester sind mit Rektifikation zum Teil komplett im Mittellauf gelandet, ohne Rektifikation nur im Schnitt so zu 60%.
Kaum Unterschiede bei den Säuren und Alkoholen.
Mit Rektifikation ist Aceton weg und leider auch a-Pinene. Diacetyl ist halbiert.
Mit Rektifikation 74 anstelle 69% Ethanolausbeute.
Also fast nur gutes bringt die Rektifikation. Ganz stark bei den Estern.
Das bedeutet auch, daß mit der Rektifikation des Vorlaufs Probleme gelöst werden können, welche bei Brennweisen, die bei anderen Aspekten, wie zum Beispiel der Menge an Säuren, Vorteile bieten, aber bei den Estern und Aldehyden nicht optimal sind. Also wenn man die Möglichkeit hat, den Vorlauf zu rektifizieren, kann man sich bei der Brennweise auf andere Dinge konzentrieren.
Neutral-Doppelbrand mit oder ohne Verdünnen zwischen den Bränden?
10l mit 10vol%.
Um ein deutliches Ergebnis zu bekommen, wird nur wenig Vorlauf abgetrennt und recht spät der Mittellauf beendet.
A)
Raubrand: bis 3l 0.1R.
Feinbrand: bis 0.03l 5R, bis 90% des Alkohols destilliert ist 5R, Vorlauf weg:
B)
Raubrand: bis 3l 0.1R, dann 3l Wasser dazu.
Feinbrand: bis 0.03l 5R, bis 90% des Alkohols destilliert ist 5R, Vorlauf weg:
Mit Wasser sind die Aldehyde etwas besser abgetrennt.
Mit Wasser sind wesentlich weniger niedrige Ester übrig, aber teilweise mehr von den besseren.
Ohne Wasser sind die höheren Alkohole etwas besser unterdrückt.
Bei den Sonstigen fällt D-Limonen auf, welches durch die Verdünnung stark in den Vorlauf gedrängt wurde, also weg ist.
Fazit: Verdünnen ist vom Geruch her neutraler und besser. Das Plus an höheren Alkoholen wird sich aber vielleicht negativ bemerkbar machen.
Turbo-Einfachbrand vs Turbo-Doppelbrand
10l mit 19vol%.
A)
Einfachbrand bis 0.05l 0.3R, dann bis 91°C 0.1R, Vorlauf weg:
B)
Raubrand bis 3.5l 0.1R.
Feinbrand bis 0.04l 0.3R, dann bis akt. 70vol% 0.1R, Vorlauf weg:
Der Doppelbrand hat 77 anstelle 49% Ethanolausbeute.
Der Doppelbrand hat etwas mehr Aldehde, vor allem aber von den guten. Im Vergleich zu Ethanol sind es allerdings etwas weniger.
Bei den Estern das gleiche.
Der Doppelbrand hat viel weniger Säuren, ist also viel weicher auf der Zunge.
Der Doppelbrand hat mehr höhere Alkohole, im Vergleich zu Ethanol aber weniger.
Fazit: Durch die sehr unterschiedliche Ethanolausbeute sind die beiden Brände sehr schlecht vergleichbar. Dadurch, daß man beim Einfachbrand so viel Ethanol verwirft, verteilen sich die flüchtigen Aromen auf weniger Alkohol. Das ist ein Vorteil vom Einfachbrand. Allerdings ist sein Säuregehalt sehr hoch, was ihn kratzig macht. Außerdem ist das Verhältnis von guten zu schlechten Stoffen innerhalb einer Stoffgruppe beim Doppelbrand meist besser.
Wenn man, um die Ausbeute beim Einfachbrand zu erhöhen, den den Mittellauf später beendet, verliert man den Vorteil des Einfachbrands, daß sich die Aromen auf weniger Alkohol verteilen, und es kommen immer mehr Säuren. Die niedrige Ausbeute rettet also den Einfachbrand. Ohne das Opfern dieses Ethanols verliert man Qualität.
Nun könnte man beim Doppelbrand meinen, daß man beim Feinbrand nicht so hohe vol% im Kessel haben sollte. Dafür muss man entweder zwischen den Bränden verdünnen oder den Raubrand verlängern.
C)
Doppelbrand wie B, aber zwischen den Bränden 1l Wasser dazu und beim Feinbrand wegen des niedrigeren Alkoholgehalts im Kessel erst bei 65vol% aktuellem Alkoholgehalt abgetrennt:
Das ist bei den Aldehyden und Alkoholen etwas schlechter und bei den Estern etwas besser. Und hat eine etwas geringere Ethanolausbeute. Bei den Säuren schaut es schlechter aus, aber bei der wichtigsten Säure, der Essigsäure, ist es sogar etwas besser.
D)
Doppelbrand wie C, aber anstelle 1l Wasser dazuzugeben wird 1l mehr raugebrannt:
Das bringt vor allem viel mehr Säuren. Und etwas mehr Ethanol und etwas mehr höhere Alkohole. Schaut nicht gut aus.
Gesamtfazit: Der Einfachbrand schaut bei den Säuren und bei der Mittellaufausbeute sehr schlecht aus. Aber die schlechte Ethanolausbeute rettet ihn in gewisser Weise. Erhöht man diese, wird er sauer. Beim Doppelbrand scheint das extrem weit herunterbrennen, um nicht allzu hohe vol% im Kessel zu haben, eine schlechte Empfehlung zu sein. Weniger weit herunterzubrennen und dann entweder verdünnt oder unverdünnt zu brennen, scheint beides besser zu sein.
Raubrand beim Doppelbrand lang oder kurz?
10l Maische mit 8vol%.
A)
Raubrand bis 2.5l 0.1R, also ein sehr kurzer Raubrand. Dann beim Feinbrand 0.03l mit 0.4R Vorlauf, Mittellauf bis 61vol% aktueller Alkoholgehalt:
B)
Raubrand bis 3.5l 0.1R, also ein langer Raubrand. Dann beim Feinbrand 0.03l mit 0.4R Vorlauf, Mittellauf bis 58vol% aktueller Alkoholgehalt. 58 anstelle 61vol% Mittellaufende, da der Alkoholgehalt des Raubrands niedriger ist:
Der kurze Raubrand hat weniger schlechte und mehr gute Aldehyde.
Und mehr von den guten Estern.
Etwas weniger Alkohole und deutlich weniger Säuren.
Er ist also sauberer trotz höherer Alkoholausbeute.
Es schaut also so aus, als bekommt man beim Doppelbrand mit langem Raubrennen mehr Geschmack, aber tendenziell etwas mehr schlechte Geschmäcker als gute.
Interessant ist vielleicht noch Phenol:
Dies ist bei dem langen Raubrand deutlich mehr vorhanden, 30.4 anstelle 22.9% im Vergleich zu Ethanol. Bei einer rauchigen Malzmaische bewirkt ein langer Raubrand also einen höheren Rauchgehalt im Whisky.
Dreifach- vs Doppelbrand
Zuerst sehr niedrigem Alkoholgehalt, 10l Maische mit 5vol%:
A)
Doppelbrand. Raubrand bis 2l mit 0.1R, dann Feinbrand 0.03l mit 0.4R Vorlauf, Mittellauf mit 0.1R bis 58vol% aktuellem Alkoholgehalt:
B)
Dreifachbrand. Raubrand bis 3l mit 0.1R, dann Mittelbrand bis 1.5l mit 0.1R, dann Feinbrand 0.03l mit 0.4R Vorlauf, Mittellauf mit 0.1R bis 65vol% aktuellem Alkoholgehalt:
Die Ethanolausbeute ist beim Dreifachbrand etwas höher, 65 anstelle 59%.
Es wurden etwas mehr schlechte Aldehyde mit dem Vorlauf abgetrennt, von den guten wurde weniger abgetrennt.
Ester wurden insgesamt weniger abgetrennt. Das betrifft aber vor allem die guten Ester.
Von den kleinen Säuren wurde viel mehr abgetrennt. Von den großen nur etwas weniger.
Es wurden im Vergleich zu Ethanol etwas mehr höhere Alkohole abgetrennt, allerdings etwas weniger Methanol.
Bei den Sonstigen Stoffen ist es unterschiedlich. Das Dreifachbrennen reduziert nicht zwingend diese Aromen. Zum Beispiel auch mit den Tails in Verbindung gebrachte Aromen wie Phenol werden nicht reduziert.
Das ist also ein klarer Sieg für den Dreifachbrand.
Ist das aber bei höherprozentiger Maische auch so?
10l Maische mit 12vol%:
C)
Doppelbrand. Raubrand bis 3.3l mit 0.1R, dann Feinbrand 0.05l mit 0.4R Vorlauf, Mittellauf mit 0.1R bis 66vol% aktuellem Alkoholgehalt:
D)
Dreifachbrand. Raubrand bis 4l mit 0.1R, dann Mittelbrand bis 2.5l mit 0.1R, dann Feinbrand 0.05l mit 0.4R Vorlauf, Mittellauf mit 0.1R bis 71vol% aktuellem Alkoholgehalt:
Die Ergebnisse sind ähnlich wie bei niedrigem Alkoholgehalt. Aber bei den Estern ist der Vorteil des Dreifachbrands verschwunden. Bei den Säuren ist der Unterschied vielleicht noch etwas größer geworden. Die Sonstigen wurden beim Dreifachbrand nun tendenziell mehr abgetrennt, was wahrscheinlich eher schlecht ist.
Das ist also kein so klarer Sieg mehr für den Dreifachbrand wie bei niedrigen vol%, aber immer noch leicht ersichtlich.
Ob der Doppelbrand irgendwann im Vorteil ist, wenn man die vol% in der Maische weiter erhöht?
10l Maische mit 19vol%:
E)
Doppelbrand. Raubrand bis 3.5l mit 0.1R, dann Feinbrand 0.05l mit 0.4R Vorlauf, Mittellauf mit 0.1R bis 72vol% aktuellem Alkoholgehalt:
F)
Dreifachbrand. Raubrand bis 4l mit 0.1R, dann Mittelbrand bis 3l mit 0.1R, dann Feinbrand 0.06l mit 0.4R Vorlauf, Mittellauf mit 0.1R bis 76vol% aktuellem Alkoholgehalt:
Der Dreifachbrand hat nur noch einen minimalen Vorteil bei den Aldehyden, bei den Estern ist er qualitativ schlechter, bei den Säuren immer noch viel besser, auch bei den Alkoholen. Die Sonstigen werden teilweise sehr stark unterdrückt. Er wird weicher als der Doppelbrand sein, aber vom Geruch schlechter.
Fazit: Es schaut so aus, als wäre der Dreifachbrand meistens besser. Allerdings schmilzt sein Vorteil, je alkoholstärker die Maische ist. Bei sehr niedrigprozentiger Maische ist er klar im Vorteil, bei Turbomaische ist es wohl Geschmackssache.
Doppelbrand vs Reflux-Einfachbrand
Dieser Vergleich ist schwer, fair durchzuführen. Denn wenn man einen Einfachbrand mit einer Refluxdestille macht, wird man den Vorlauf wahrscheinlich mit viel Reflux abtrennen. Diese Option hat man mit einer normalen Potstill aber nicht. Für einen Vergleich aber ist es interessant, wenn die Refluxdestille testweise zuerst mal auf diese Möglichkeit verzichtet:
10l mit 10vol%.
A)
Potstill-Doppelbrand, Raubrand 3l mit 0.1R, Feinbrand Vorlauf 0.05l mit 0.4R und dann bis 65vol% aktuellem Alkoholgehalt mit 0.1R:
B)
Reflux Einfachbrand, Feinbrand Vorlauf 0.05l mit 0.8R und dann versucht den Alkoholgehalt nicht unter 70vol% sinken zu lassen, bis man 64% des Alkohols als Mittellauf hat. Das ist das Protokoll:
Der Doppelbrand hat wesentlich mehr Aldehyde, auch von den schlechten, aber vor allem von den guten.
Er hat wesentlich mehr Ester, ebenfalls auch von den schlechten, aber vor allem von den guten.
Er hat weniger Essigsäure, aber mehr von den größeren Säuren. Da Essigsäure den Löwenanteil ausmacht, ist er etwas weicher.
Bei den Alkoholen sind beide Brennweisen ähnlich, der Doppelbrand hat etwas mehr Methanol.
Bei den Sonstigen gibt es ein paar, wo der Doppelbrand deutlich besser abschneidet.
Der Doppelbrand hat also qualitative Vorzüge, braucht aber wahrscheinlich eine etwas großzügigere Vorlaufabtrennung, um mehr von den kleinen Aldehyden und Estern abzutrennen.
Aber dieser Vergleich war ja unfair, weil die Refluxdestille auf ihre Möglichkeit, den Vorlauf zu rektifizieren, verzichtet hat.
Deswegen nun
C)
das gleiche Einfachbrand-Protokoll aber mit 0.02l Vorlauf mit 3R:
Die Probleme mit den Aldehyden und den Estern sind nun gelöst. Auch das Aceton ist weg. Das Problem mit der Essigsäure bleibt aber.
Ein Nachteil ist die nun etwas erhöhte Menge an höheren Alkoholen.
Der Reflux-Einfachbrand mit viel Reflux beim Vorlauf ist also besser in der Nase, der Potstill-Doppelbrand aber ist weicher.
Wenn man nun beim Doppelbrand ebenfalls den Vorlauf rektifizieren kann, bekommt man best of both worlds:
D)
Wie A, aber Vorlauf 0.02l mit 3R:
Es sind sehr viele der großen Aldehyde übrig, obwohl die kleinen fast weg sind. Ethylacetat wurde weniger gut abgetrennt als bei C, aber die größeren Ester wurden mehr geschont. Vielleicht hätte man 0.03 anstelle 0.02l Vorlauf eingeben sollen. Auch hier ist der Nachteil, daß die höheren Alkohole im Mittellauf etwas ansteigen, wenn man den Vorlauf rektifiziert. Die Vorteile bei den Säuren bleiben. Der Doppelbrand mit Rektifizierung des Vorlaufs ist also ähnlich gut oder vielleicht sogar besser in der Nase als der Reflux-Einfachbrand mit Rektifizierung des Vorlaufs und er ist weicher als der Reflux-Einfachbrand.
Raubrand mit Rektifikation anstelle Rau- und Mittelbrand?
10l mit 8vol%.
A)
Raubrand bis 3l 0.1R, Mittelbrand bis 1.5l 0.1R:
B)
Reflux-Raubrand bis 1.5l 52.7vol%. Dafür sind 0.63R nötig:
Bei beiden Versionen sind so gut wie alle Aldehyde, Ester, Alkohole und Sonstige im Destillat. Beim Doppelbrand ein bisschen vollständiger, was tendenziell etwas besser ausschaut. Aber bei den Säuren gibt es große Unterschiede. Der Doppelbrand unterdrückt die Essigsäure und Ameisensäure besser, der Einfachbrand eher die größeren Säuren. Da die Essigsäure am Häufigsten ist, wird der Doppelbrand etwas weicher sein. Außerdem hat er mehr von den interessanten Säuren mit höherem Potential, welche beim Feinbrand dann vielleicht wertvolle Ester bilden.
Nun könnte man den Refluxbrand aber auch mit ansteigendem Reflux machen, die Alkoholstärke also nie unter einen bestimmten Punkt fallen lassen. Hier das Protokoll:
C)
Das Endergebnis:
Die Ergebnisse von A, B und C bilden Linien. Bei Essigsäure ist A zum Beispiel bei 3.7%, B bei 4.4% und C nun bei 4.7%. Oder Phenol ist bei A 67.8%, bei B 55.9% und bei C nun 46.4%.
Das ist für mich überraschend. Ich dachte, C würde bezüglich der Säuren sauberer sein. Hinsichtlich der wichtigen Essigsäure ist aber das Gegenteil passiert.
Wenn man B also schlechter als A findet (was wahrscheinlich ist), wird man C noch schlechter finden.
Edit: Inzwischen wird in diesen wie auch in anderen Rechnern nicht mehr die Zahl "Rektifikation" eingegeben, sondern die Anzahl der theoretischen Böden. Eine ideale Potstill hat 1 theoretischen Boden. Das hat vorher Rektifikation 0 bedeutet. Also theoretische Böden = Rektifikation +1.
Näheres dazu hier: Theoretische Böden
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Theoretische Böden
Wir haben einen neuen Rechner:
Theoretische Böden
Er berechnet aus den Temperaturen oder Alkoholstärken im Kessel und im Dampf die Anzahl der theoretischen Böden, also die Anzahl der stattfindenden Destillationsstufen.
Wer unsere Rechner kennt, fragt sich nun vielleicht, warum wir nicht die Zahl Rektifikation berechnen lassen, welche ja im Prinzip das gleiche bedeutet (zum Beispiel 1.25 theoretische Böden bedeuten 0.25 Rektifikation, also theoretische Böden = Rektifikation + 1) und welche man in andere Rechner eingeben kann.
Das liegt einfach daran, daß wir diese Bezeichnung in allen Rechnern durch "theoretische Böden" ersetzen wollen. Denn theoretische Böden ist der wissenschaftlich korrekte und übliche Begriff dafür, welcher hier im Forum aber normalerweise falsch verwendet wird. Auch in manchen Rechnern wie den McCabe-Thiele-Rechnern und auch im Glossar wird dieser Begriff falsch verwendet oder definiert.
Wir verwenden den Begriff normalerweise, um Packungskolonnen in einen Vergleich mit Bödenkolonnen setzen zu können. Also wie schreiben, eine Destille hat 10 theoretische Böden, wenn ihre Rektifikationsleistung der einer Kolonne mit 10 realen Böden entspricht.
Die korrekte Definition ist allerdings eine andere:
theoretische Böden sind die, welche übrigbleiben, wenn man die Effizienz berücksichtigt. Und die Effizienz hängt vor allem vom Reflux ab.
https://en.wikipedia.org/wiki/Theoretical_plate
Dies betrifft auch den Begriff HETP (height equivalent to a theoretical plate, Höhenequivalent eines theoretischen Bodens), welcher die Kolonnenlänge bezeichnet, welche man für einen theoretischen Boden benötigt.
theoretische Böden = Höhe / HETP
Wenn aber die theoretischen Böden abhängig von der Effizienz und damit vom Reflux sind, also man bei 0% Reflux nur 0 theoretische Böden von der Kolonne bekommt, ist das HETP dieser Kolonne also HETP = Höhe / 0 = unendlich.
Also wenn wir den Begriff HETP im Forum verwenden, meinen wir normalerweise das minimale HETP, also den Wert, wenn theor. Böden = reale Böden (und die Böden einer Packungskolonne sind nunmal genauso real wie die einer Plattenkolonne).
Also, die (hoffentlich) korrekten Definitionen:
Platten, Böden, oder reale Platten, reale Böden:
Entweder die wirklich optisch abzählbaren Böden einer Plattenkolonne oder das dieser Leistung entsprechende bei einer Packungskolonne.
theoretische Böden oder theoretische Platten oder theoretische Bodenzahl:
Das, was im konkreten Betrieb einer Destille an Rektifikationsleistung erreicht oder benötigt wird. Diesen Begriff kann man auch bei einer Potstill, also einer Destille mit nur passivem Reflux, verwenden.
HETP:
Die Kolonnenlänge, die einem theoretischem Boden entspricht.
Gleichbedeutend sind die Begriffe "Bodenhöhe", "theoretische Bodenhöhe" und "Trennstufenhöhe", in Formeln dann meist als H abgekürzt.
minimales HETP:
Die Kolonnenlänge, die bei 100% Reflux und auch sonstigen idealen Bedingungen einem theoretischen Boden entspricht.
Destillationsstufen (distillation stages oder distillation steps):
Dieser Begriff bezeichnet die Stufen, die man in Siedediagramme einzeichnen kann. Aber manche Siedediagramme berücksichtigen die Effizienz (McCabe-Thiele-Diagramme), andere nicht (die Diagramme mit der Siedelinse). Der Begriff kann also reale oder theoretische Böden bezeichnen. Deswegen sollte man diesen Begriff nur verwenden, wenn es um ein konkretes Diagramm geht.
Ich werde das Glossar, die Rechner und meine Rechnervorstellungen in nächster Zeit dementsprechend korrigieren.
Theoretische Böden
Er berechnet aus den Temperaturen oder Alkoholstärken im Kessel und im Dampf die Anzahl der theoretischen Böden, also die Anzahl der stattfindenden Destillationsstufen.
Wer unsere Rechner kennt, fragt sich nun vielleicht, warum wir nicht die Zahl Rektifikation berechnen lassen, welche ja im Prinzip das gleiche bedeutet (zum Beispiel 1.25 theoretische Böden bedeuten 0.25 Rektifikation, also theoretische Böden = Rektifikation + 1) und welche man in andere Rechner eingeben kann.
Das liegt einfach daran, daß wir diese Bezeichnung in allen Rechnern durch "theoretische Böden" ersetzen wollen. Denn theoretische Böden ist der wissenschaftlich korrekte und übliche Begriff dafür, welcher hier im Forum aber normalerweise falsch verwendet wird. Auch in manchen Rechnern wie den McCabe-Thiele-Rechnern und auch im Glossar wird dieser Begriff falsch verwendet oder definiert.
Wir verwenden den Begriff normalerweise, um Packungskolonnen in einen Vergleich mit Bödenkolonnen setzen zu können. Also wie schreiben, eine Destille hat 10 theoretische Böden, wenn ihre Rektifikationsleistung der einer Kolonne mit 10 realen Böden entspricht.
Die korrekte Definition ist allerdings eine andere:
theoretische Böden sind die, welche übrigbleiben, wenn man die Effizienz berücksichtigt. Und die Effizienz hängt vor allem vom Reflux ab.
https://en.wikipedia.org/wiki/Theoretical_plate
Also auch eine Kolonne mit richtigen Böden hat theoretische Böden.Since an actual, physical plate can never be a 100% efficient equilibrium stage, the number of actual plates is more than the required theoretical plates.
Na = Nt / E
where Na is the number of actual, physical plates or trays, Nt is the number of theoretical plates or trays and E is the plate or tray efficiency.
Dies betrifft auch den Begriff HETP (height equivalent to a theoretical plate, Höhenequivalent eines theoretischen Bodens), welcher die Kolonnenlänge bezeichnet, welche man für einen theoretischen Boden benötigt.
theoretische Böden = Höhe / HETP
Wenn aber die theoretischen Böden abhängig von der Effizienz und damit vom Reflux sind, also man bei 0% Reflux nur 0 theoretische Böden von der Kolonne bekommt, ist das HETP dieser Kolonne also HETP = Höhe / 0 = unendlich.
Also wenn wir den Begriff HETP im Forum verwenden, meinen wir normalerweise das minimale HETP, also den Wert, wenn theor. Böden = reale Böden (und die Böden einer Packungskolonne sind nunmal genauso real wie die einer Plattenkolonne).
Also, die (hoffentlich) korrekten Definitionen:
Platten, Böden, oder reale Platten, reale Böden:
Entweder die wirklich optisch abzählbaren Böden einer Plattenkolonne oder das dieser Leistung entsprechende bei einer Packungskolonne.
theoretische Böden oder theoretische Platten oder theoretische Bodenzahl:
Das, was im konkreten Betrieb einer Destille an Rektifikationsleistung erreicht oder benötigt wird. Diesen Begriff kann man auch bei einer Potstill, also einer Destille mit nur passivem Reflux, verwenden.
HETP:
Die Kolonnenlänge, die einem theoretischem Boden entspricht.
Gleichbedeutend sind die Begriffe "Bodenhöhe", "theoretische Bodenhöhe" und "Trennstufenhöhe", in Formeln dann meist als H abgekürzt.
minimales HETP:
Die Kolonnenlänge, die bei 100% Reflux und auch sonstigen idealen Bedingungen einem theoretischen Boden entspricht.
Destillationsstufen (distillation stages oder distillation steps):
Dieser Begriff bezeichnet die Stufen, die man in Siedediagramme einzeichnen kann. Aber manche Siedediagramme berücksichtigen die Effizienz (McCabe-Thiele-Diagramme), andere nicht (die Diagramme mit der Siedelinse). Der Begriff kann also reale oder theoretische Böden bezeichnen. Deswegen sollte man diesen Begriff nur verwenden, wenn es um ein konkretes Diagramm geht.
Ich werde das Glossar, die Rechner und meine Rechnervorstellungen in nächster Zeit dementsprechend korrigieren.
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Destillen-Druckberechnung
Wir haben ein kleines neues Tool unter "Sonstige und praxisferne Berechnungen":
Destillen-Druckberechnung
Es berechnet aus der Alkoholstärke und der Siedetemperatur den Luftdruck. Diesen vergleicht er dann mit dem eingegebenen Umgebungsluftdruck. Er kann also berechnen, ob und wie viel Über- oder Unterdruck es in der Destille gibt, ohne daß man den Druck messen muss.
Dies kann er sowohl mit der Alkoholstärke und Temperatur im Kessel als auch im Dampf beziehungsweise Destillat.
So eine Berechnung hat natürlich aber nur einen Sinn, wenn eine hohe Genauigkeit der Messungen gewährleistet ist.
Destillen-Druckberechnung
Es berechnet aus der Alkoholstärke und der Siedetemperatur den Luftdruck. Diesen vergleicht er dann mit dem eingegebenen Umgebungsluftdruck. Er kann also berechnen, ob und wie viel Über- oder Unterdruck es in der Destille gibt, ohne daß man den Druck messen muss.
Dies kann er sowohl mit der Alkoholstärke und Temperatur im Kessel als auch im Dampf beziehungsweise Destillat.
So eine Berechnung hat natürlich aber nur einen Sinn, wenn eine hohe Genauigkeit der Messungen gewährleistet ist.
-
Hügelwilli
- Site Admin
- Beiträge: 835
- Registriert: 10. Okt 2018, 21:47
Rumrechner
Wir haben einen neuen Rechner: Rumrechner
Es geht um die Zutaten von Rummaischen. Ähnlich wie der Obstmaischerechner 2 berechnet auch dieser Rechner den Einfluss von unvergärbaren Stoffen auf Dichte-Messwerte der Maische.
Eine zu hohe Dichte verursacht durch entweder zu viel Zucker oder zu viele unvergärbare Stoffe ist ja ein häufiges Problem bei Rummaischen. Und die allgemeine Verunsicherung, wenn man aus den "zu hohen" Messwerten nichts ablesen kann, verhindert leicht die korrekte Einschätzung des Zustands der Gärung und verursacht falsche Entscheidungen.
In der Anmerkung unter dem Rechner stehen noch viele Details.
Es geht um die Zutaten von Rummaischen. Ähnlich wie der Obstmaischerechner 2 berechnet auch dieser Rechner den Einfluss von unvergärbaren Stoffen auf Dichte-Messwerte der Maische.
Eine zu hohe Dichte verursacht durch entweder zu viel Zucker oder zu viele unvergärbare Stoffe ist ja ein häufiges Problem bei Rummaischen. Und die allgemeine Verunsicherung, wenn man aus den "zu hohen" Messwerten nichts ablesen kann, verhindert leicht die korrekte Einschätzung des Zustands der Gärung und verursacht falsche Entscheidungen.
In der Anmerkung unter dem Rechner stehen noch viele Details.