Mit "Begleitstoffen" sind hier flüchtige aromatische chemische Verbindungen gemeint, welche, obwohl sie auf die Menge bezogen nur einen kleinen Teil ausmachen, den Geschmack der Spirituosen bestimmen.
Nicht gemeint sind nach der Destillation zugesetzte Stoffe wie zum Beispiel Zucker oder Fruchsäfte.
Wie sich diese Stoffe bei der Destillation verhalten, hat einen wesentlichen Einfluss auf das geschmackliche Endergebnis. Die entscheidende Rolle spielt dabei deren Flüchtigkeit. Es gibt die absolute Flüchtigkeit und Flüchtigkeiten relativ zu der Flüchtigkeit anderer Stoffe. Wenn im Dampf der Anteil des Begleitstoffs höher ist als in der Flüssigkeit, dann ist die absolute Flüchtigkeit größer als 1. Wenn er niedriger ist, dann ist die abs. Flüchtigkeit niedriger als 1. In wissenschaftlichen Schriften bezieht sich diese Zahl immer auf das Molverhältnis. Also wenn der Stoff in der Flüssigkeit einen Molanteil von 0.0001 hat und im Dampf 0.001, dann ist seine Flüchtigkeit 10. In unseren Rechnern wird dagegen meist die Flüchtigkeit bezogen auf die Masse verwendet, da diese anschaulicher ist. Also wenn der Stoff in der Flüssigkeit 0.01gew% hat und im Dampf 0.1gew%, dann ist seine Flüchtigkeit 10.
Die Flüchtigkeit hängt neben den individuellen Eigenschaften des Stoffs sehr stark von der Alkoholstärke ab. Grundsätzlich sind alle Aromastoffe umso flüchtiger, je weniger Alkohol sich in der Lösung befindet. Hier ein Beispiel: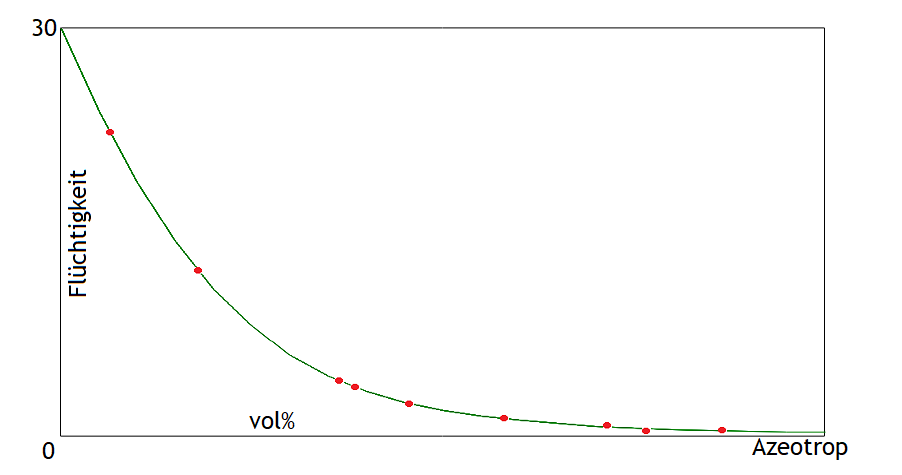 Das ist die Kurve von Isobutanol.
Die roten Punkte sind die Messwerte, die grüne Linie die daraus abgeleitete Kurve.
Das ist die Kurve von Isobutanol.
Die roten Punkte sind die Messwerte, die grüne Linie die daraus abgeleitete Kurve.
Bei den anderen Stoffen schaut die Kurve sehr ähnlich aus: Immer sinkt sie von links nach rechts und immer hängt sie nach unten durch. Die abs. Flüchtigkeit liegt je nach Alkoholstärke bei Isobutanol bezogen auf den Molanteil zwischen 0.38 und 30. Das bedeutet, bei einer Destillation ohne Alkohol ist anteilig im Dampf die 30-fache Isobutanol-Stoffmenge wie in der Flüssigkeit. Und bei einer Destillation mit azeotropem Alkohol in der Destille, ist anteilig im Dampf fast nur 1/3 der Stoffmenge Isobutanol wie in der Flüssigkeit. Folglich hat die Alkoholstärke bei der Destillation einen riesigen Einfluss auf den Geschmack.
Die Messdaten gehen davon aus, daß die Begleitstoffe in so niedriger Konzentration vorliegen, daß deren Moleküle sich nicht gegenseitig beeinflussen. Das ist bei Destillationen mit hoher Rektifikation aber nicht mehr der Fall. Wenn man mit diesen Daten starke Rektifikationen berechnet, werden daher für den Anfang der Destillation und später für den fast schlagartigen Übergang zwischen Azeotrop und 0% Alkohol im Destillat, zu extreme Aufkonzentrierungen der Begleitstoffe ausgegeben. Hier liegt also eine Einschränkung für alle unsere die Begleitstoffe betreffenden Rechner. Die generellen Aussagen der Rechner, also zum Beispiel, ob sich ein Stoff im Vor- oder im Nachlauf konzentriert, bleiben dann aber trotzdem gültig. Jedoch sind die Konzentrierungen in der Praxis dann längst nicht so extrem, wie es die Rechner berechnen.
Auch die Flüchtigkeitskurve von Ethanol schaut so aus. Die Flüchtigkeit ist bei (fast) 0vol% ca. 11 und beim Azeotrop genau 1. Für unsere Überlegungen ist meist der Verlauf der relativen Flüchtigkeit zu der von Ethanol interessant. Hat der Begleitstoff bei einer bestimmten Alkoholstärke eine abs. Flüchtigkeit von 10 und Ethanol bei derselben Alkohlstärke die abs. Flüchtigkeit 2, ist die relative Flüchtigkeit des Begleitstoffs bei dieser Alkoholstärke 10 / 2 = 5.
An dem Verlauf der relativen Flüchtigkeit der Stoffe kann man ihr sehr unterschiedliches Verhalten bei der Destillation erkennen. Also z.B. ob sie im Vorlauf oder Nachlauf landen.
Alleine anhand der Kurven kann man aber nur sehen, wie an einem bestimmten Startpunkt einer Destillation sich die Begleitstoffe verhalten. Nicht gut kann man spätere Zeitpunkte vorhersagen, da es schwer einzuschätzen ist, wie viel bei einem späteren Zeitpunkt bei dann höherer Flüchtigkeit, da der Alkoholgehalt gesunken ist, noch von dem Stoff in der Destille ist. Also einerseits hat man dann die höhere Flüchtigkeit andererseits aber nicht mehr den gleichen Anteil des Begleitstoffs in der Destille. Das kann nur ein Rechenprogramm, welches die Destillation in Kleinstschritten durchrechnet.
Es kann also durchaus sein, daß, obwohl seine Flüchtigkeit steigt, immer weniger des Stoffs ins Destillat kommt. Bei allen typischen Vorlaufstoffen ist das so. Typische Vorlaufstoffe sind die, welche über den ganzen Alkoholstärkenbereich eine höhere Flüchtigkeit als Ethanol haben; also überall eine relative Flüchtigkeit höher als 1. Typische Nachlaufstoffe haben über den ganzen Bereich eine niedrigere Flüchtigkeit als Ethanol; also überall eine relative Flüchtigkeit niedriger als 1. Und Stoffe mit niedrigerer Flüchtigkeit als Wasser bleiben mehr oder weniger in der Destille zurück. Kompliziert ist es bei Stoffen, welche bei niedriger Alkoholstärke flüchtiger sind als Ethanol und bei hoher Alkohlstärke weniger flüchtig. Dann hängt es von der Anfangsalkoholstärke und der Rektifikation ab, ob der Stoff eher im Vor- oder eher im Nachlauf landet. Bei höheren Alkoholen ist das der Fall.
In den Rechnern sind die Begleitstoffe in Gruppen geordnet. Innerhalb jeder Gruppe sind sie nach ihrer Molmasse geordnet. Die Molmasse korreliert oft mit "extremerem" Verhalten: eine höhere Molmasse bedeutet meist einer größere Spanne der Flüchtigkeit zwischen 0vol% Alkohol und dem Azeotrop und ein stärkeres Aroma. In mehreren der Stoffgruppen gibt es aber jeweils einen Stoff mit eher geringer Molmasse, der mengenmäßig die anderen bei weitem übertrifft. Hier ist es dann sinnvoll die relative Flüchtigkeit der anderen Stoffe aus dieser Stoffgruppe zu diesem Hauptstoff zu berechnen, da dies Aussagen über die Qualität der Spirituose zulässt.
Wie sich diese Stoffe bei der Destillation verhalten, hat einen wesentlichen Einfluss auf das geschmackliche Endergebnis. Die entscheidende Rolle spielt dabei deren Flüchtigkeit. Es gibt die absolute Flüchtigkeit und Flüchtigkeiten relativ zu der Flüchtigkeit anderer Stoffe. Wenn im Dampf der Anteil des Begleitstoffs höher ist als in der Flüssigkeit, dann ist die absolute Flüchtigkeit größer als 1. Wenn er niedriger ist, dann ist die abs. Flüchtigkeit niedriger als 1. In wissenschaftlichen Schriften bezieht sich diese Zahl immer auf das Molverhältnis. Also wenn der Stoff in der Flüssigkeit einen Molanteil von 0.0001 hat und im Dampf 0.001, dann ist seine Flüchtigkeit 10. In unseren Rechnern wird dagegen meist die Flüchtigkeit bezogen auf die Masse verwendet, da diese anschaulicher ist. Also wenn der Stoff in der Flüssigkeit 0.01gew% hat und im Dampf 0.1gew%, dann ist seine Flüchtigkeit 10.
Die Flüchtigkeit hängt neben den individuellen Eigenschaften des Stoffs sehr stark von der Alkoholstärke ab. Grundsätzlich sind alle Aromastoffe umso flüchtiger, je weniger Alkohol sich in der Lösung befindet. Hier ein Beispiel:
Bei den anderen Stoffen schaut die Kurve sehr ähnlich aus: Immer sinkt sie von links nach rechts und immer hängt sie nach unten durch. Die abs. Flüchtigkeit liegt je nach Alkoholstärke bei Isobutanol bezogen auf den Molanteil zwischen 0.38 und 30. Das bedeutet, bei einer Destillation ohne Alkohol ist anteilig im Dampf die 30-fache Isobutanol-Stoffmenge wie in der Flüssigkeit. Und bei einer Destillation mit azeotropem Alkohol in der Destille, ist anteilig im Dampf fast nur 1/3 der Stoffmenge Isobutanol wie in der Flüssigkeit. Folglich hat die Alkoholstärke bei der Destillation einen riesigen Einfluss auf den Geschmack.
Die Messdaten gehen davon aus, daß die Begleitstoffe in so niedriger Konzentration vorliegen, daß deren Moleküle sich nicht gegenseitig beeinflussen. Das ist bei Destillationen mit hoher Rektifikation aber nicht mehr der Fall. Wenn man mit diesen Daten starke Rektifikationen berechnet, werden daher für den Anfang der Destillation und später für den fast schlagartigen Übergang zwischen Azeotrop und 0% Alkohol im Destillat, zu extreme Aufkonzentrierungen der Begleitstoffe ausgegeben. Hier liegt also eine Einschränkung für alle unsere die Begleitstoffe betreffenden Rechner. Die generellen Aussagen der Rechner, also zum Beispiel, ob sich ein Stoff im Vor- oder im Nachlauf konzentriert, bleiben dann aber trotzdem gültig. Jedoch sind die Konzentrierungen in der Praxis dann längst nicht so extrem, wie es die Rechner berechnen.
Auch die Flüchtigkeitskurve von Ethanol schaut so aus. Die Flüchtigkeit ist bei (fast) 0vol% ca. 11 und beim Azeotrop genau 1. Für unsere Überlegungen ist meist der Verlauf der relativen Flüchtigkeit zu der von Ethanol interessant. Hat der Begleitstoff bei einer bestimmten Alkoholstärke eine abs. Flüchtigkeit von 10 und Ethanol bei derselben Alkohlstärke die abs. Flüchtigkeit 2, ist die relative Flüchtigkeit des Begleitstoffs bei dieser Alkoholstärke 10 / 2 = 5.
An dem Verlauf der relativen Flüchtigkeit der Stoffe kann man ihr sehr unterschiedliches Verhalten bei der Destillation erkennen. Also z.B. ob sie im Vorlauf oder Nachlauf landen.
Alleine anhand der Kurven kann man aber nur sehen, wie an einem bestimmten Startpunkt einer Destillation sich die Begleitstoffe verhalten. Nicht gut kann man spätere Zeitpunkte vorhersagen, da es schwer einzuschätzen ist, wie viel bei einem späteren Zeitpunkt bei dann höherer Flüchtigkeit, da der Alkoholgehalt gesunken ist, noch von dem Stoff in der Destille ist. Also einerseits hat man dann die höhere Flüchtigkeit andererseits aber nicht mehr den gleichen Anteil des Begleitstoffs in der Destille. Das kann nur ein Rechenprogramm, welches die Destillation in Kleinstschritten durchrechnet.
Es kann also durchaus sein, daß, obwohl seine Flüchtigkeit steigt, immer weniger des Stoffs ins Destillat kommt. Bei allen typischen Vorlaufstoffen ist das so. Typische Vorlaufstoffe sind die, welche über den ganzen Alkoholstärkenbereich eine höhere Flüchtigkeit als Ethanol haben; also überall eine relative Flüchtigkeit höher als 1. Typische Nachlaufstoffe haben über den ganzen Bereich eine niedrigere Flüchtigkeit als Ethanol; also überall eine relative Flüchtigkeit niedriger als 1. Und Stoffe mit niedrigerer Flüchtigkeit als Wasser bleiben mehr oder weniger in der Destille zurück. Kompliziert ist es bei Stoffen, welche bei niedriger Alkoholstärke flüchtiger sind als Ethanol und bei hoher Alkohlstärke weniger flüchtig. Dann hängt es von der Anfangsalkoholstärke und der Rektifikation ab, ob der Stoff eher im Vor- oder eher im Nachlauf landet. Bei höheren Alkoholen ist das der Fall.
In den Rechnern sind die Begleitstoffe in Gruppen geordnet. Innerhalb jeder Gruppe sind sie nach ihrer Molmasse geordnet. Die Molmasse korreliert oft mit "extremerem" Verhalten: eine höhere Molmasse bedeutet meist einer größere Spanne der Flüchtigkeit zwischen 0vol% Alkohol und dem Azeotrop und ein stärkeres Aroma. In mehreren der Stoffgruppen gibt es aber jeweils einen Stoff mit eher geringer Molmasse, der mengenmäßig die anderen bei weitem übertrifft. Hier ist es dann sinnvoll die relative Flüchtigkeit der anderen Stoffe aus dieser Stoffgruppe zu diesem Hauptstoff zu berechnen, da dies Aussagen über die Qualität der Spirituose zulässt.
Die Stoffgruppen und jeweiligen Vergleichsstoffe:
Aldehyde, Hauptstoff Acetaldehyd:
Aldehyde sind sehr flüchtige, leichte Gerüche. Je weiter unten in der Tabelle, umso intensiver und komplexer ist das Aroma der Aldehyde. Ein gutes Aroma haben Aldehyde erst ab Valeraldehyd oder vielleicht schon ab Butanal nach unten. Aldehyde sind meist sehr flüchtige Stoffe. Die Flüchtigkeit relativ zu Ethanol ist meist sehr hoch. Daher finden sie sich meist im Vorlauf wieder. Acetaldehyd ist bei weitem das häufigste Aldehyd und ein unangenehmer, eindimensionaler Aromastoff. Mit hoher Rektifikation ist es möglich, das Acetaldehyd im Vorlauf zu konzentriert abzutrennen und aber höherwertige Aldehyde teilweise zu behalten.
Ester, Hauptstoff Ethylacetat:
Ester sind fruchtige Aromen mit tendenziell hoher Flüchtigkeit. Der bei weitem häufigste Ester Ethylacetat mit seiner niedrigen Molmasse ist aber eher ein unangenehmer, eindimensionaler Lösungsmittelgeruch. Je weiter unten in der Tabelle, umso intensiver, komplexer und besser ist das Aroma der Ester. Schon der zweite Stoff von oben hat eine weitaus bessere Qualität als das Ethylacetat. Mit hoher Rektifikation ist es möglich, das Ethylacetat im Vorlauf zu konzentriert abzutrennen und aber höherwertige Ester teilweise zu behalten. Manche sehr hochwertige Ester brauchen niedrige Alkoholstärke und keine oder nur wenig Rektifikation, um nicht im Kessel zurückzubleiben.
Säuren, Hauptstoff Essigsäure:
Säuren sind grundsätzlich unangenehme Begleitstoffe. Die bei weitem häufigste ist die Essigsäure. Diese ist hauptverantwortlich dafür, daß der Nachlauf ein harsches Mundgefühl verursacht. Wenn ein Destillat nicht weich ist, liegt es meist an ihr. Die Säuren mit höherer Molmasse sind vom Geruch her extrem unangenehm. Allgemein ist also ein Destillat mit möglichst wenig Säuren besser. Mit Rektifikation lassen sich Säuren sehr gut mit dem Nachlauf bzw der Schlempe abtrennen. Wenn man die anderen Säuren relativ zur Essigsäure im Mittellauf reduziert, hat das zwar theoretisch positive Auswirkungen, aber meist fällt es wenig ins Gewicht. Allerdings können aus Säuren Ester gebildet werden. Und die Säuren weiter unten in der Liste bilden die interessanteren Ester als die Säuren weiter oben. Also kann es durchaus sinnvoll sein, bei der ersten Destillation ein eher die unteren Säuren mitzunehmen, um deren Veresterung mehr Gelegenheiten zu geben, und dann erst bei der letzten Destillation zu versuchen, allgemein die Säuren zu reduzieren. Das ist allerdings nicht so möglich, wie man das vielleicht gerne hätte: Beendet man den Raubrand früher, hat der Raubrand allgemein weniger Säuren und aber eher viel der Säuren mit hoher Molmasse. Beim Feinbarnd wird man also wenige, aber tendenziell hochwertigere Ester bekommen. Außerdem wird man wahlweise entweder ein sehr weiches Destillat bekommen oder eine hohe Mittellaufausbeute. Will man aber ein sehr esterhaltiges Destillat bekommen, muss man den Raubrand sehr weit herunterbrennen und hat dann erstens vor allem niederwertige Ester, die man mit dem Vorlauf gut abtrennen muss, und zweitens einen Säuregehalt, der entweder den Brand sehr unweich macht oder ein sehr frühes Mittellaufende und damit eine schlechte Mittellaufausbeute erzwingt. Und das ist relativ unabhängig von der Anfangsalkoholstärke oder der Rektifikation. Der Weg zu einem komplexen Brand mit vielen und höherwertigen Estern muss daher schon vorher bei der Gärung eingeschlagen werden. Daß man also dort Organismen fördert, welche höherwertige Säuren produzieren.
Alkohole, der Hauptstoff ist Ethanol selber:
Der einzige geniesbare Alkohol mit geringer Molmasse ist Ethanol. Methanol riecht ähnlich wie Ethanol. Die anderen mehr oder weniger nach Lösungsmittel oder fuselig. Zwar sind auch hier die höherwertigen Stoffe weiter unten, aber nur die untersten drei können als positiv angesehen werden. Also sollte man bei der Destillation die anderen Alkohole eher versuchen abzutrennen. Je nach Destillationsweise kann das bei den Alkoholen im Vorlauf oder im Nachlauf sein. Je höher die Alkoholstärke oder die Rektifikaton, desto eher sammeln sich die Alkohole im Nachlauf. Außer beim Methanol, wo es andersherum ist. Denn es ist der einzige Alkohol mit kleinerer Molmasse als Ethanol. Aber genauso wie bei den Säuren können die Alkohole verestern und die Alkohole weiter unten bilden die interessanteren Ester, weswegen es sinnvoll sein kann, die Alkohole erst beim Feinbrand abzutrennen. Wobei man auch hier eher schon bei der Gärung das Fundament für höherwertige Alkohole legen sollte.
Sonstigen Stoffe, kein Hauptstoff:
Sie sind auch nach ihrer Molmasse geordnet, was hier aber keine Bedeutung hat. Aceton hat ganz klar kein gutes Aroma, spielt aber bei unseren Destillaten keine große Rolle. Bei Diacetyl ist es eine Geschmacksfrage und eine Frage der Menge. Phenol ist eher nur interessant für rauchige Destillate. Die anderen Stoffe sind positiv. Allerdings sind es spezielle Stoffe mit Citrusaromen, welche in den meisten unserer Brände gar nicht merkbar auftauchen. Aber sie gehören zur Gruppe der Terpene, welche ganz vielfältige Aromen haben können und in aromatischen Destillaten oft eine große Rolle spielen. Also können diese Werte durchaus auch für Brände ohne Citrusaromen interessant sein.
Aldehyde, Hauptstoff Acetaldehyd:
Aldehyde sind sehr flüchtige, leichte Gerüche. Je weiter unten in der Tabelle, umso intensiver und komplexer ist das Aroma der Aldehyde. Ein gutes Aroma haben Aldehyde erst ab Valeraldehyd oder vielleicht schon ab Butanal nach unten. Aldehyde sind meist sehr flüchtige Stoffe. Die Flüchtigkeit relativ zu Ethanol ist meist sehr hoch. Daher finden sie sich meist im Vorlauf wieder. Acetaldehyd ist bei weitem das häufigste Aldehyd und ein unangenehmer, eindimensionaler Aromastoff. Mit hoher Rektifikation ist es möglich, das Acetaldehyd im Vorlauf zu konzentriert abzutrennen und aber höherwertige Aldehyde teilweise zu behalten.
Ester, Hauptstoff Ethylacetat:
Ester sind fruchtige Aromen mit tendenziell hoher Flüchtigkeit. Der bei weitem häufigste Ester Ethylacetat mit seiner niedrigen Molmasse ist aber eher ein unangenehmer, eindimensionaler Lösungsmittelgeruch. Je weiter unten in der Tabelle, umso intensiver, komplexer und besser ist das Aroma der Ester. Schon der zweite Stoff von oben hat eine weitaus bessere Qualität als das Ethylacetat. Mit hoher Rektifikation ist es möglich, das Ethylacetat im Vorlauf zu konzentriert abzutrennen und aber höherwertige Ester teilweise zu behalten. Manche sehr hochwertige Ester brauchen niedrige Alkoholstärke und keine oder nur wenig Rektifikation, um nicht im Kessel zurückzubleiben.
Säuren, Hauptstoff Essigsäure:
Säuren sind grundsätzlich unangenehme Begleitstoffe. Die bei weitem häufigste ist die Essigsäure. Diese ist hauptverantwortlich dafür, daß der Nachlauf ein harsches Mundgefühl verursacht. Wenn ein Destillat nicht weich ist, liegt es meist an ihr. Die Säuren mit höherer Molmasse sind vom Geruch her extrem unangenehm. Allgemein ist also ein Destillat mit möglichst wenig Säuren besser. Mit Rektifikation lassen sich Säuren sehr gut mit dem Nachlauf bzw der Schlempe abtrennen. Wenn man die anderen Säuren relativ zur Essigsäure im Mittellauf reduziert, hat das zwar theoretisch positive Auswirkungen, aber meist fällt es wenig ins Gewicht. Allerdings können aus Säuren Ester gebildet werden. Und die Säuren weiter unten in der Liste bilden die interessanteren Ester als die Säuren weiter oben. Also kann es durchaus sinnvoll sein, bei der ersten Destillation ein eher die unteren Säuren mitzunehmen, um deren Veresterung mehr Gelegenheiten zu geben, und dann erst bei der letzten Destillation zu versuchen, allgemein die Säuren zu reduzieren. Das ist allerdings nicht so möglich, wie man das vielleicht gerne hätte: Beendet man den Raubrand früher, hat der Raubrand allgemein weniger Säuren und aber eher viel der Säuren mit hoher Molmasse. Beim Feinbarnd wird man also wenige, aber tendenziell hochwertigere Ester bekommen. Außerdem wird man wahlweise entweder ein sehr weiches Destillat bekommen oder eine hohe Mittellaufausbeute. Will man aber ein sehr esterhaltiges Destillat bekommen, muss man den Raubrand sehr weit herunterbrennen und hat dann erstens vor allem niederwertige Ester, die man mit dem Vorlauf gut abtrennen muss, und zweitens einen Säuregehalt, der entweder den Brand sehr unweich macht oder ein sehr frühes Mittellaufende und damit eine schlechte Mittellaufausbeute erzwingt. Und das ist relativ unabhängig von der Anfangsalkoholstärke oder der Rektifikation. Der Weg zu einem komplexen Brand mit vielen und höherwertigen Estern muss daher schon vorher bei der Gärung eingeschlagen werden. Daß man also dort Organismen fördert, welche höherwertige Säuren produzieren.
Alkohole, der Hauptstoff ist Ethanol selber:
Der einzige geniesbare Alkohol mit geringer Molmasse ist Ethanol. Methanol riecht ähnlich wie Ethanol. Die anderen mehr oder weniger nach Lösungsmittel oder fuselig. Zwar sind auch hier die höherwertigen Stoffe weiter unten, aber nur die untersten drei können als positiv angesehen werden. Also sollte man bei der Destillation die anderen Alkohole eher versuchen abzutrennen. Je nach Destillationsweise kann das bei den Alkoholen im Vorlauf oder im Nachlauf sein. Je höher die Alkoholstärke oder die Rektifikaton, desto eher sammeln sich die Alkohole im Nachlauf. Außer beim Methanol, wo es andersherum ist. Denn es ist der einzige Alkohol mit kleinerer Molmasse als Ethanol. Aber genauso wie bei den Säuren können die Alkohole verestern und die Alkohole weiter unten bilden die interessanteren Ester, weswegen es sinnvoll sein kann, die Alkohole erst beim Feinbrand abzutrennen. Wobei man auch hier eher schon bei der Gärung das Fundament für höherwertige Alkohole legen sollte.
Sonstigen Stoffe, kein Hauptstoff:
Sie sind auch nach ihrer Molmasse geordnet, was hier aber keine Bedeutung hat. Aceton hat ganz klar kein gutes Aroma, spielt aber bei unseren Destillaten keine große Rolle. Bei Diacetyl ist es eine Geschmacksfrage und eine Frage der Menge. Phenol ist eher nur interessant für rauchige Destillate. Die anderen Stoffe sind positiv. Allerdings sind es spezielle Stoffe mit Citrusaromen, welche in den meisten unserer Brände gar nicht merkbar auftauchen. Aber sie gehören zur Gruppe der Terpene, welche ganz vielfältige Aromen haben können und in aromatischen Destillaten oft eine große Rolle spielen. Also können diese Werte durchaus auch für Brände ohne Citrusaromen interessant sein.
Der in Maischen normalerweise sehr niedrige pH-Wert erhöht die Flüchtigkeit von Säuren.
Das bedeutet, in der Praxis werden bei Raubränden die Säuren etwas früher als berechnet im Destillat landen.
Beim Destillieren finden chemische Reaktionen statt. Manche Moleküle verbinden sich, andere trennen sich. Dies können die Rechner nicht berechnen. Zum Beispiel findet meistens eine merkbare Veresterung statt. Bei dieser verbinden sich Säuren und Alkohole zu Estern. Es verschwinden also Säuren und Alkohole aus der Rechnung und es kommen Ester hinzu.
Wegen diesen und wahrscheinlich auch noch weiteren Unterschieden zwischen Theorie und Praxis eignen sich die Rechner, welche unsere Begleitstoffdaten verwenden, weniger für absolute Berechnungen, sondern eher für vergleichende. Das bedeutet, man kann weniger einen konkreten Abtrennpunkt berechnen lassen, sondern eher Szenarien vergleichen und dann allgemeine Aussagen treffen. So lassen sich Antworten zu vielen wichtigen Fragen finden. Hier Beispiele unter der Verwendung unserer Rechner:
Die Reihenfolge ist zufällig.
Beim Destillieren finden chemische Reaktionen statt. Manche Moleküle verbinden sich, andere trennen sich. Dies können die Rechner nicht berechnen. Zum Beispiel findet meistens eine merkbare Veresterung statt. Bei dieser verbinden sich Säuren und Alkohole zu Estern. Es verschwinden also Säuren und Alkohole aus der Rechnung und es kommen Ester hinzu.
Wegen diesen und wahrscheinlich auch noch weiteren Unterschieden zwischen Theorie und Praxis eignen sich die Rechner, welche unsere Begleitstoffdaten verwenden, weniger für absolute Berechnungen, sondern eher für vergleichende. Das bedeutet, man kann weniger einen konkreten Abtrennpunkt berechnen lassen, sondern eher Szenarien vergleichen und dann allgemeine Aussagen treffen. So lassen sich Antworten zu vielen wichtigen Fragen finden. Hier Beispiele unter der Verwendung unserer Rechner:
Ist Methanol im Vorlauf?
Es ist inzwischen allgemein bekannt, daß sich Methanol nicht im Vorlauf konzentriert.
Z.B. mit dem Begleitstoffesimulator 1 kann man es nachprüfen:
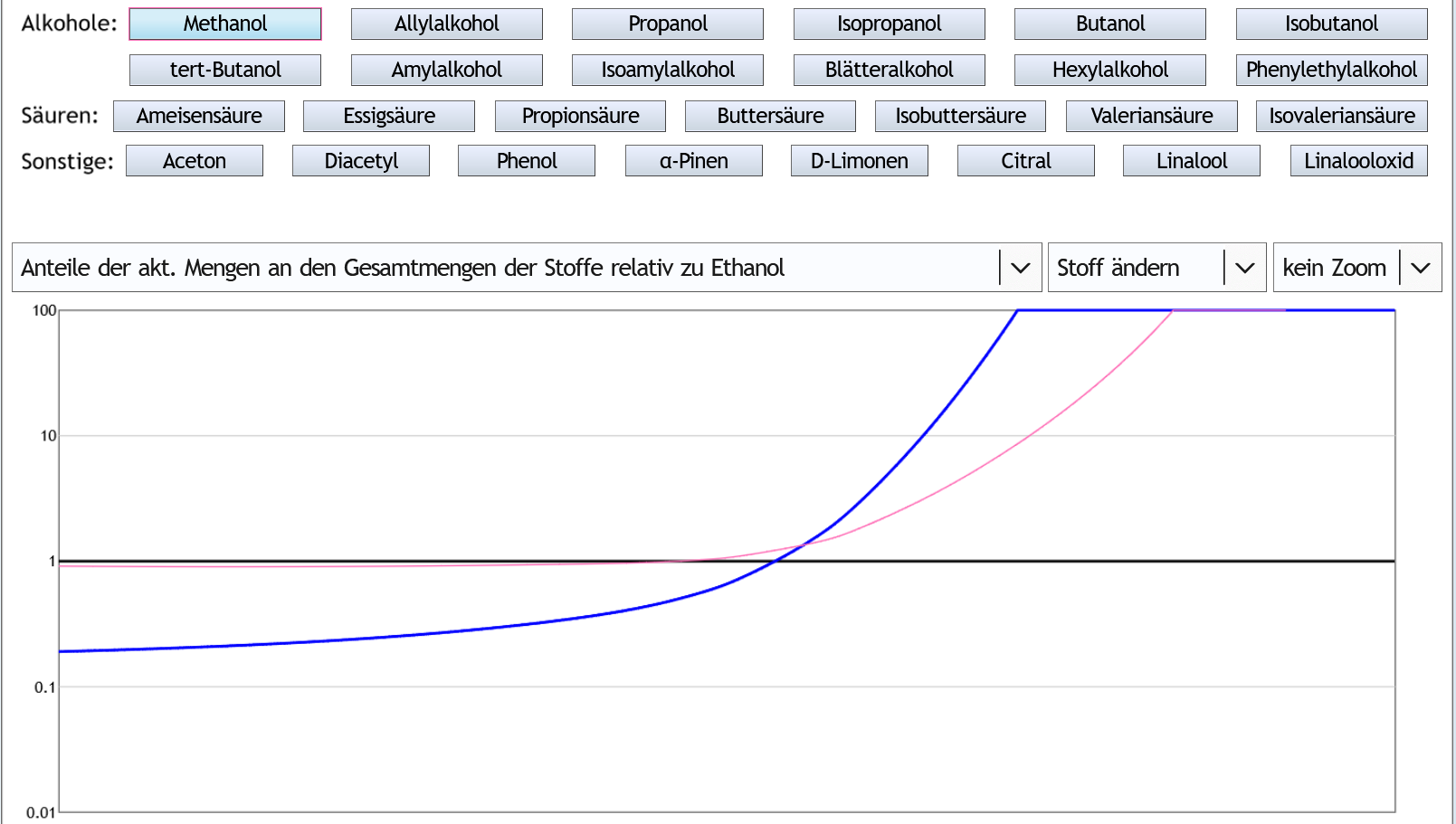
5vol% in der Destille, 1 theor. Boden, Zoom x3: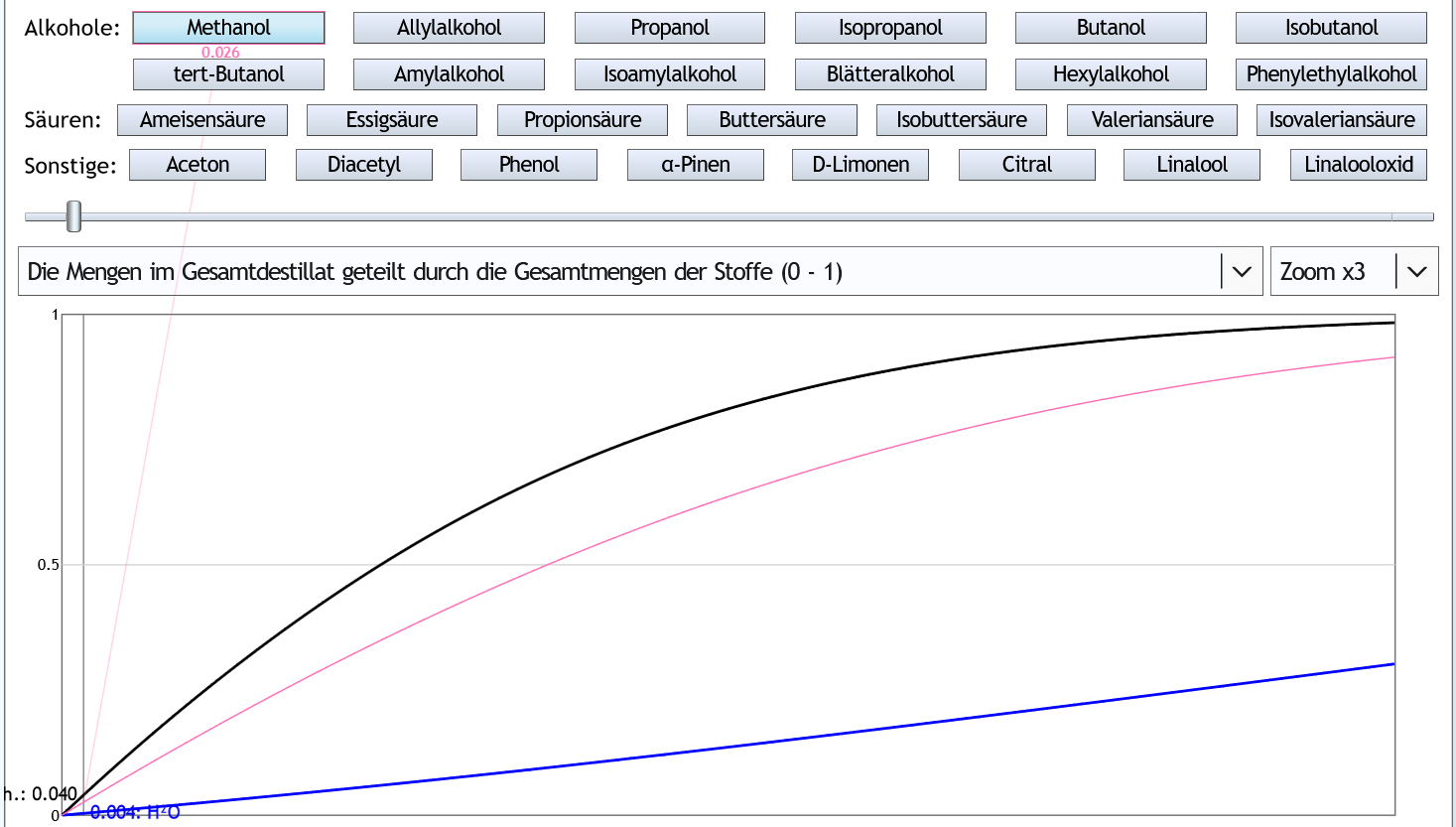 Die Kurve von Methanol (rosa) ist unter der von Ethanol (schwarz).
Methanol geht also langsamer ins Destillat als Ethanol.
Wenn man 4% (0.04) des Ethanols als Vorlauf abtrennt, hat man nur 2.6% (0.026) des Methanols entfernt.
Die Kurve von Methanol (rosa) ist unter der von Ethanol (schwarz).
Methanol geht also langsamer ins Destillat als Ethanol.
Wenn man 4% (0.04) des Ethanols als Vorlauf abtrennt, hat man nur 2.6% (0.026) des Methanols entfernt.
Wenn man diesen Raubrand nach 25% des Destilleninhalts beendet: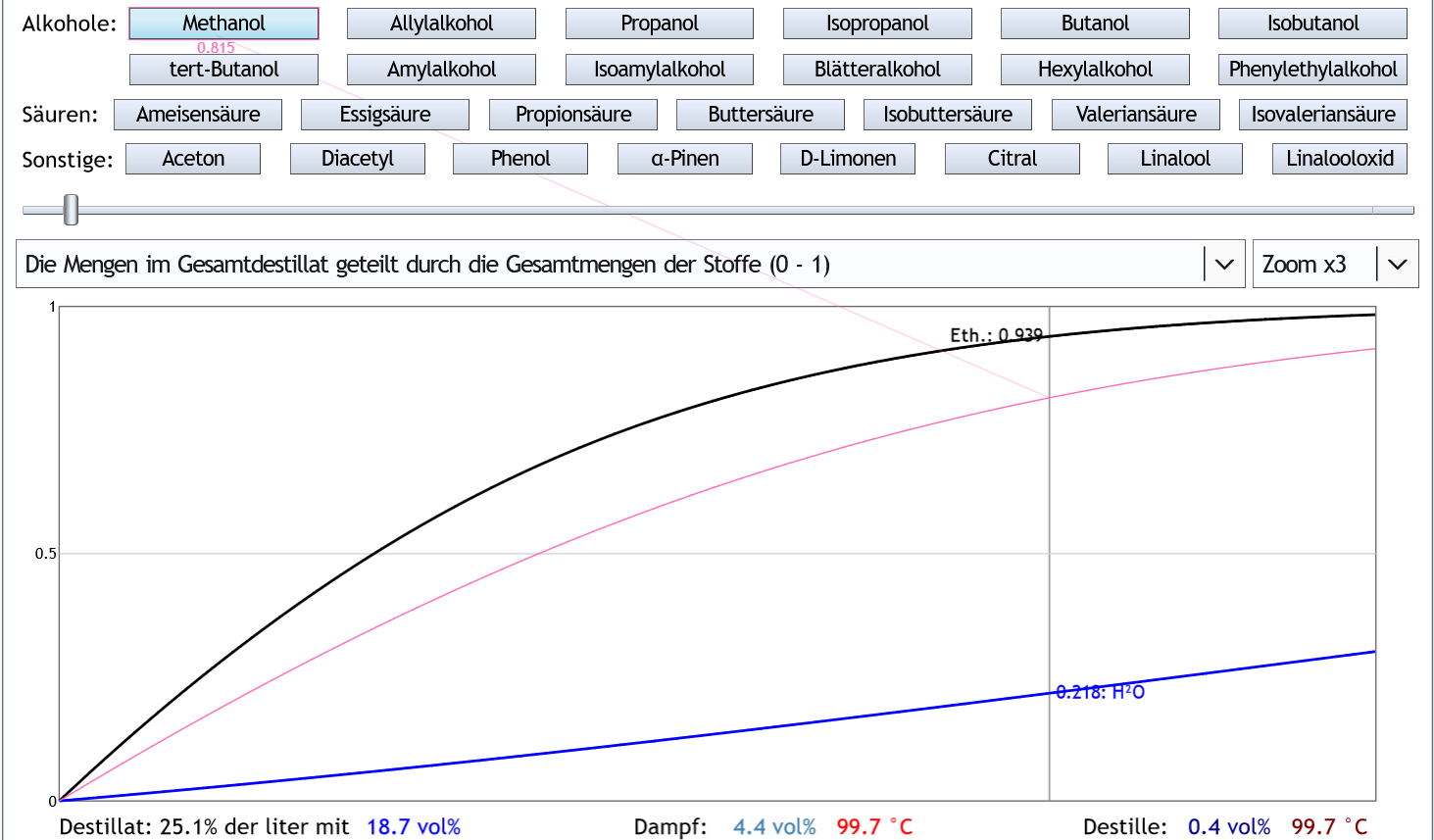 Dann hat man 93.9% des Ethanols und 81.5% des Methanols gesammelt.
Das ist ein Verhältnis von
Dann hat man 93.9% des Ethanols und 81.5% des Methanols gesammelt.
Das ist ein Verhältnis von 81.5 / 93.9 = 0.87.
Wenn man dagegen den Raubrand erst nach 33% des Destilleninhalts beendet: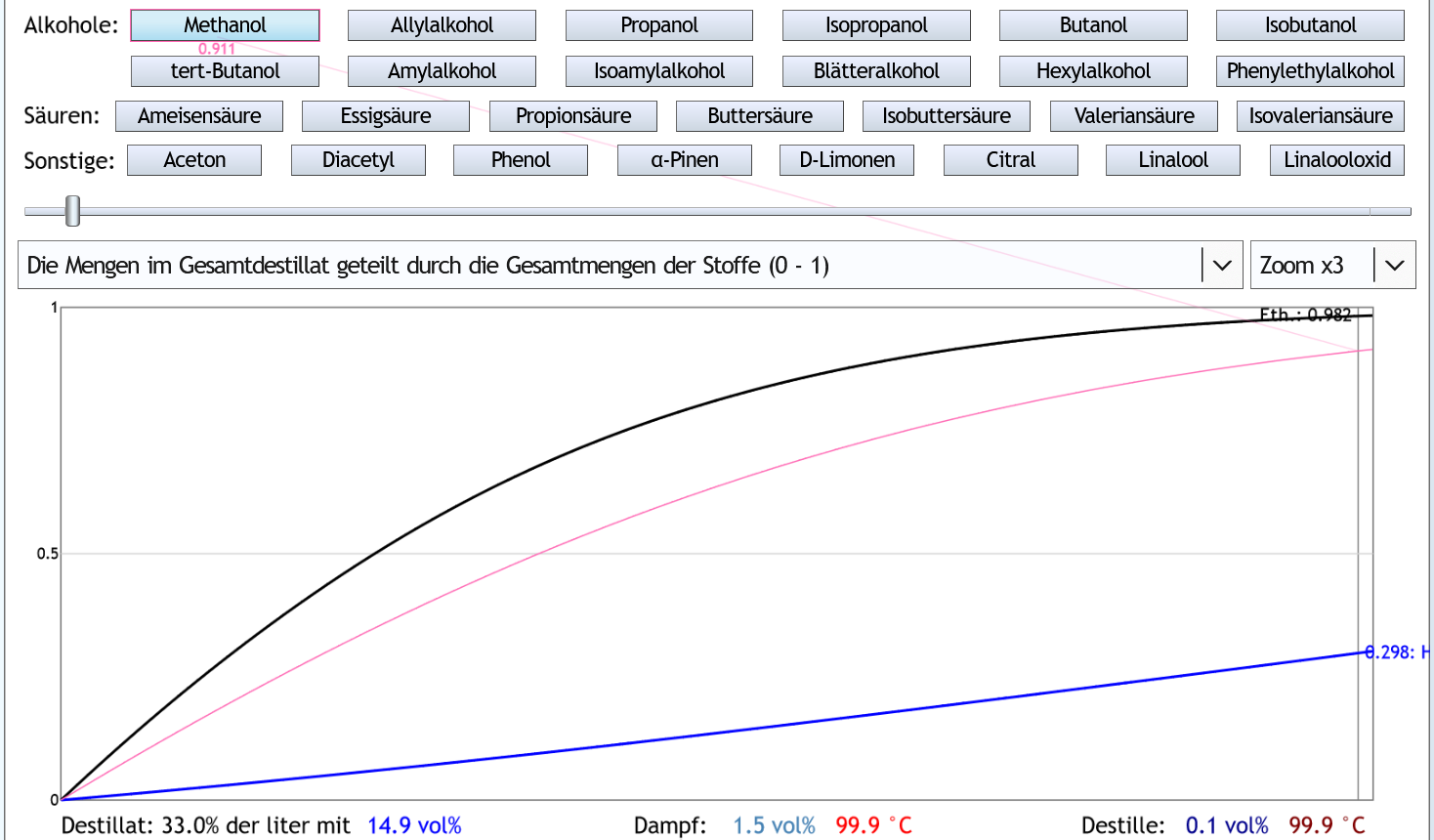 Dann hat man 98.2% des Ethanols und 91.1% des Methanols gesammelt.
Das ist ein Verhältnis von 0.93.
Das längere Sammeln hat also den Methanolanteil erhöht.
Allerdings nur in geringem Umfang.
Dann hat man 98.2% des Ethanols und 91.1% des Methanols gesammelt.
Das ist ein Verhältnis von 0.93.
Das längere Sammeln hat also den Methanolanteil erhöht.
Allerdings nur in geringem Umfang.
Bzw. in anderer Darstellungsweise: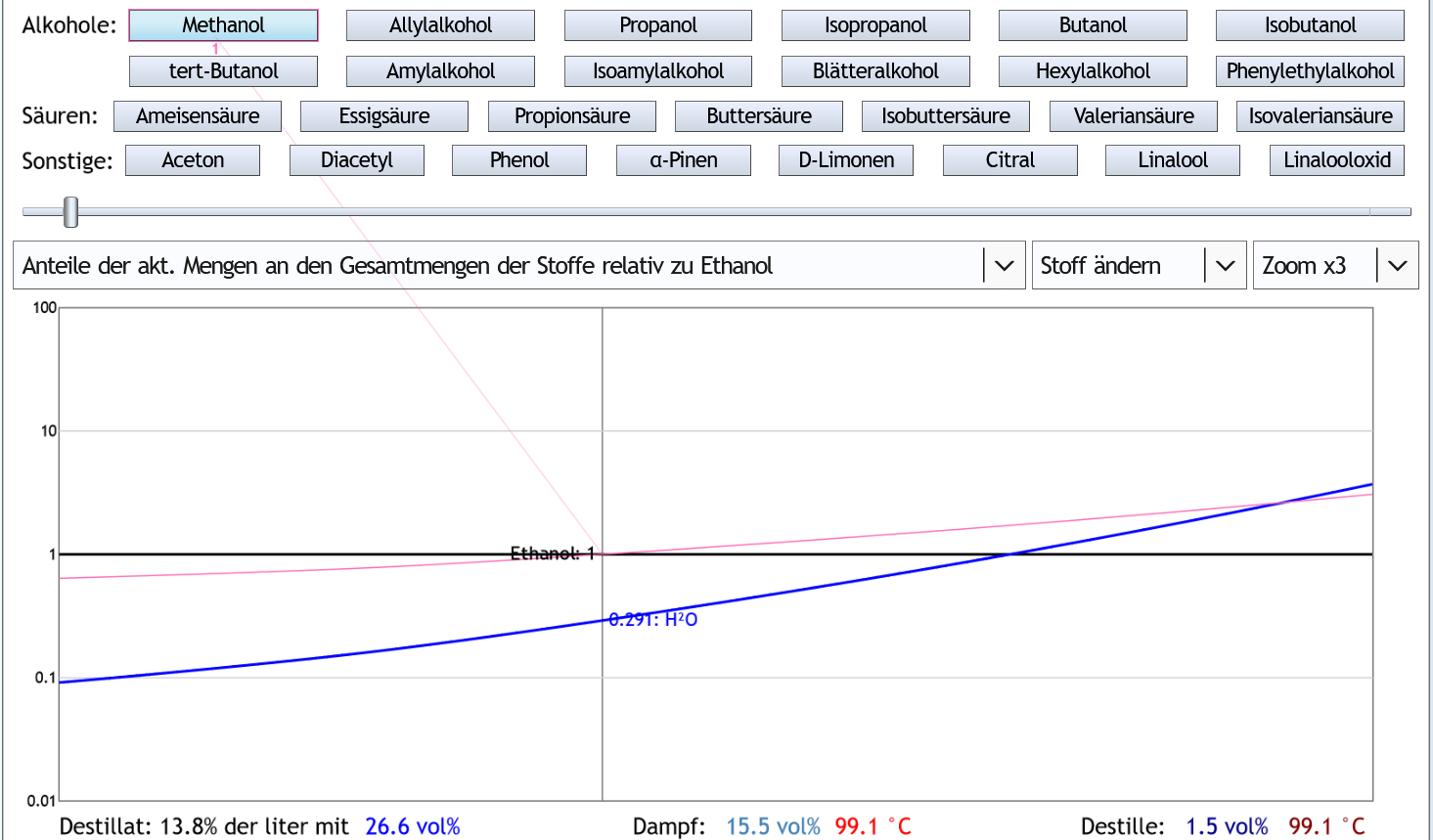 Alles Destillat, bevor man 13.8% des Destilleninhalts destilliert hat (Schnittpunkt rosa und dickere schwarze Linie), verringert schlussendlich im Schnapsglas den Methanolanteil;
alles danach erhöht ihn wieder.
Also Vorlauf abzutrennen erhöht den Methanolanteil etwas.
Und weit runterbrennen erhöht den Methanolanteil ebenfalls etwas.
Eine geringfügige Reduzierung des Methanols bekommt man in diesem Fall (Raubrand ohne Rektifikation mit niedrigen vol% im Kessel) also, wenn man keinen Vorlauf abtrennt und nicht weit herunterbrennt.
Alles Destillat, bevor man 13.8% des Destilleninhalts destilliert hat (Schnittpunkt rosa und dickere schwarze Linie), verringert schlussendlich im Schnapsglas den Methanolanteil;
alles danach erhöht ihn wieder.
Also Vorlauf abzutrennen erhöht den Methanolanteil etwas.
Und weit runterbrennen erhöht den Methanolanteil ebenfalls etwas.
Eine geringfügige Reduzierung des Methanols bekommt man in diesem Fall (Raubrand ohne Rektifikation mit niedrigen vol% im Kessel) also, wenn man keinen Vorlauf abtrennt und nicht weit herunterbrennt.
Nun mit 40vol% im Kessel: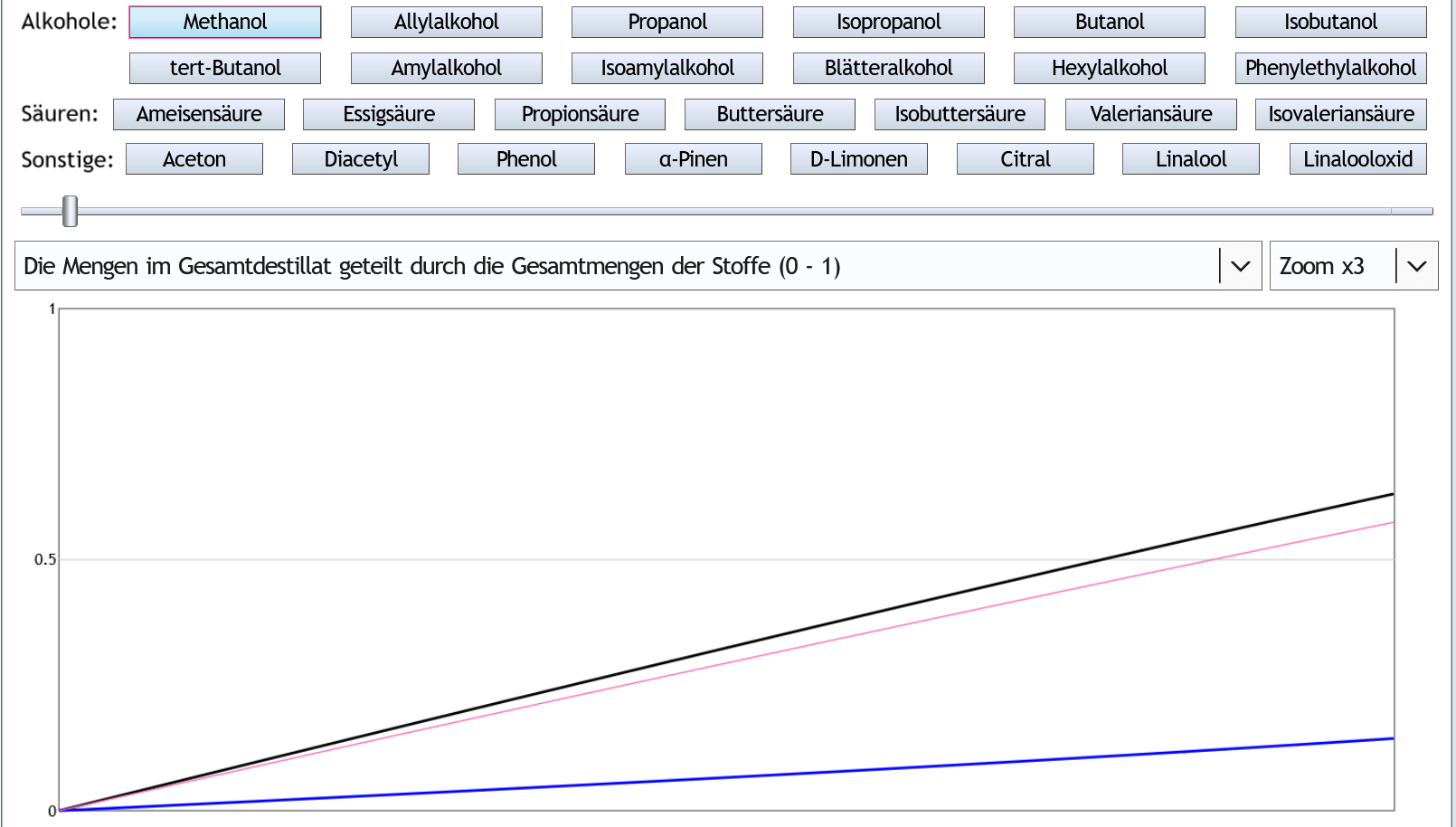
 Das ist sehr ähnlich verglichen mit 5vol% in der Destille.
Nur verhält sich Methanol noch deutlich ähnlicher zu Ethanol, wodurch Abtrennen des Vor- oder Nachlaufs noch weniger Auswirkungen auf den Methanolanteil hat.
Das ist sehr ähnlich verglichen mit 5vol% in der Destille.
Nur verhält sich Methanol noch deutlich ähnlicher zu Ethanol, wodurch Abtrennen des Vor- oder Nachlaufs noch weniger Auswirkungen auf den Methanolanteil hat.
Nun mit Rektifikation:
5vol%, 6 theor. Böden, Zoom x10: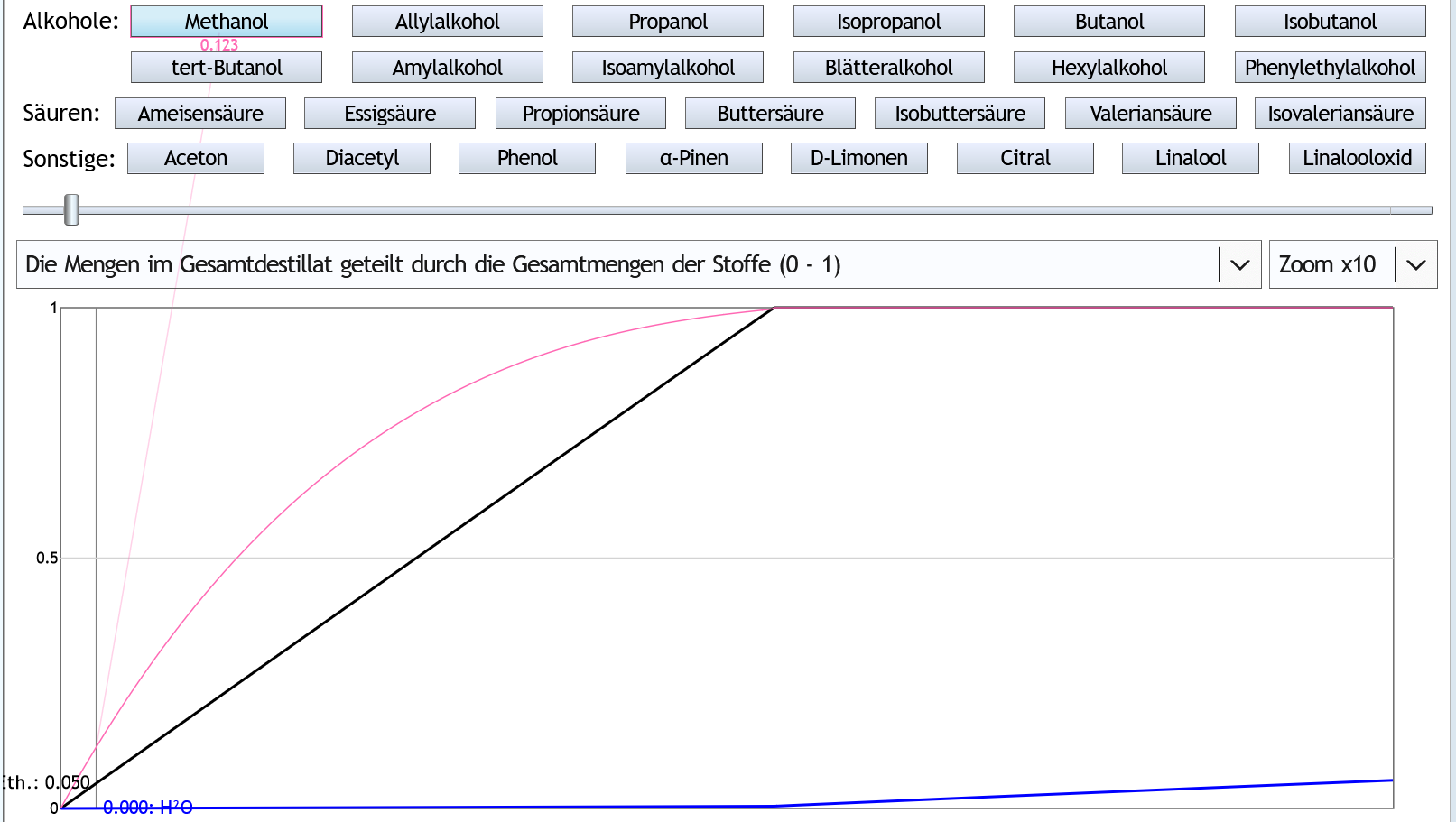 Durch die Rektifikation konzentriert sich das Methanol etwas im Vorlauf.
Wenn man 5% (0.05) des Ethanols als Vorlauf abtrennt, ist man aber trotzdem nur 12.3% (0.123) des Methanols losgeworden.
Durch die Rektifikation konzentriert sich das Methanol etwas im Vorlauf.
Wenn man 5% (0.05) des Ethanols als Vorlauf abtrennt, ist man aber trotzdem nur 12.3% (0.123) des Methanols losgeworden.
Nun 40vol%, 6 theor. Böden, Zoom x10: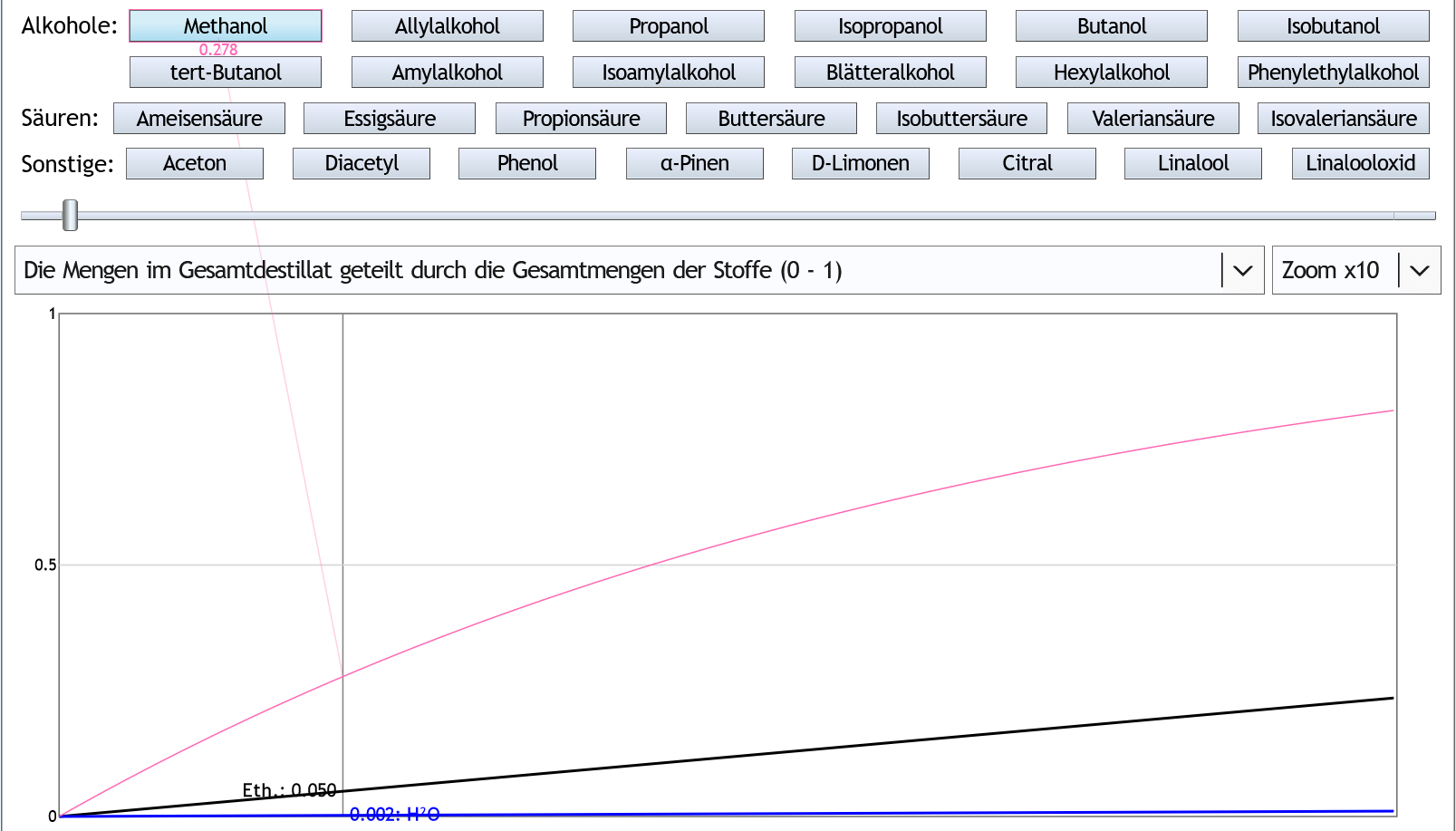 Hier wird man mit dem Vorlaufschnitt nach 5% des Ethanols 27.8% des Methanols los.
Hier wird man mit dem Vorlaufschnitt nach 5% des Ethanols 27.8% des Methanols los.
Es ist also nicht wirklich effektiv das alles. Man bekommt auch mit einer kleineren Refluxdestille das meiste nicht los.
Wenn man nun noch höhere Prozente in die Destille füllt und mehr Rektifikation einsetzt, behauptet der Rechner, kann man das Methanol relativ schnell ziemlich komplett abtrennen:
95vol%, 10 theor. Böden, Zoom x10: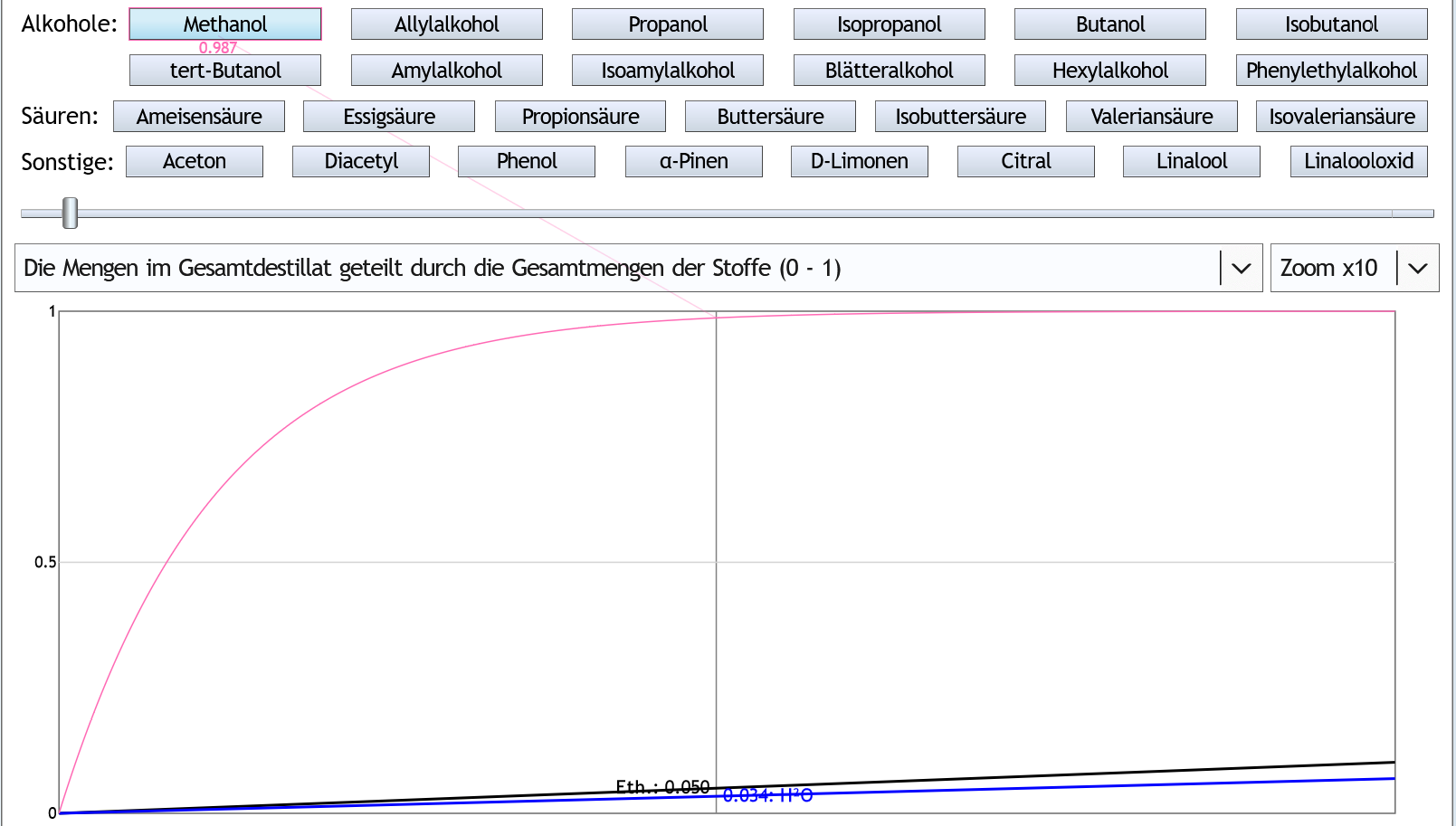 Nach 5% des Ethanols hat man 98.7% des Methanols abgetrennt.
Und so macht es auch die Industrie.
Nicht die Schnapsindustrie, sondern die für Industriealkohol.
Wir können das im Prinzip auch.
In der Praxis wird es allerdings weniger gut klappen;
erstens, da die richtigen Vorlaufstoffe (welche zum Teil sehr gute Aromastoffe sind) sich "vordrängeln" werden, wir so also höchstens Neutralalkohol machen können;
und zweitens dann der Effekt ins Spiel kommt, daß sich die Moleküle der Stoffe gegenseitig beeinflussen, die Abtrennung also nicht so scharf ist, wie mit diesen Daten berechnet.
Außerdem entsteht Methanol ja eigentlich nur bei der Vergärung von pektinhaltigem Obst.
Und das möchte man ja meist ohne oder mit nur wenig Rektifikation brennen.
Höchstens, um aus den Vor- und Nachläufen der Obst-Feinbrände Neutralalkohol zu machen, ist das interessant.
Und man sollte sich gut überlegen, ob man sich damit sicher fühlt, so hohe Alkoholstärken in die Destille zu geben.
Nach 5% des Ethanols hat man 98.7% des Methanols abgetrennt.
Und so macht es auch die Industrie.
Nicht die Schnapsindustrie, sondern die für Industriealkohol.
Wir können das im Prinzip auch.
In der Praxis wird es allerdings weniger gut klappen;
erstens, da die richtigen Vorlaufstoffe (welche zum Teil sehr gute Aromastoffe sind) sich "vordrängeln" werden, wir so also höchstens Neutralalkohol machen können;
und zweitens dann der Effekt ins Spiel kommt, daß sich die Moleküle der Stoffe gegenseitig beeinflussen, die Abtrennung also nicht so scharf ist, wie mit diesen Daten berechnet.
Außerdem entsteht Methanol ja eigentlich nur bei der Vergärung von pektinhaltigem Obst.
Und das möchte man ja meist ohne oder mit nur wenig Rektifikation brennen.
Höchstens, um aus den Vor- und Nachläufen der Obst-Feinbrände Neutralalkohol zu machen, ist das interessant.
Und man sollte sich gut überlegen, ob man sich damit sicher fühlt, so hohe Alkoholstärken in die Destille zu geben.
5vol% in der Destille, 1 theor. Boden, Zoom x3:
Wenn man diesen Raubrand nach 25% des Destilleninhalts beendet:
Wenn man dagegen den Raubrand erst nach 33% des Destilleninhalts beendet:
Bzw. in anderer Darstellungsweise:
Nun mit 40vol% im Kessel:
Nun mit Rektifikation:
5vol%, 6 theor. Böden, Zoom x10:
Nun 40vol%, 6 theor. Böden, Zoom x10:
Es ist also nicht wirklich effektiv das alles. Man bekommt auch mit einer kleineren Refluxdestille das meiste nicht los.
Wenn man nun noch höhere Prozente in die Destille füllt und mehr Rektifikation einsetzt, behauptet der Rechner, kann man das Methanol relativ schnell ziemlich komplett abtrennen:
95vol%, 10 theor. Böden, Zoom x10:
Sind die höheren Alkohole im Nachlauf?
Höhere Alkohole (oft auch Fuselalkohole genannt) werden meist als Nachlaufstoffe bezeichnet.
Das ist aber nur teilweise richtig.
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 1 berechnet.
5vol%, 1 theor. Boden: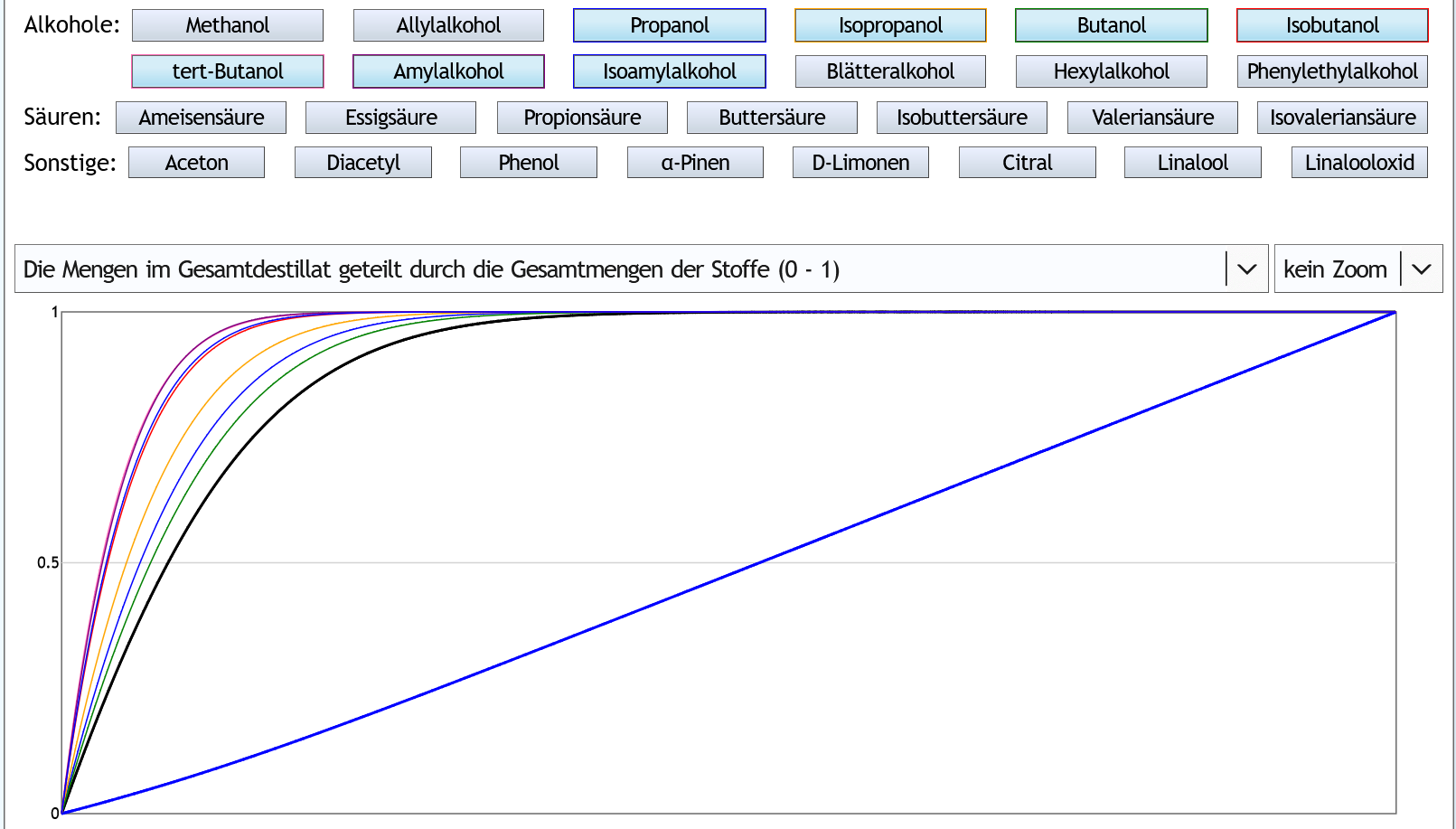 Die typischen und häufigsten höheren Alkohole (dünne bunte Linien) gehen alle schneller ins Destillat über als Ethanol (dickere schwarze Linie).
Es gibt dadurch eine geringfügige Anreicherung im Vorlauf.
Die typischen und häufigsten höheren Alkohole (dünne bunte Linien) gehen alle schneller ins Destillat über als Ethanol (dickere schwarze Linie).
Es gibt dadurch eine geringfügige Anreicherung im Vorlauf.
Andere Darstellung: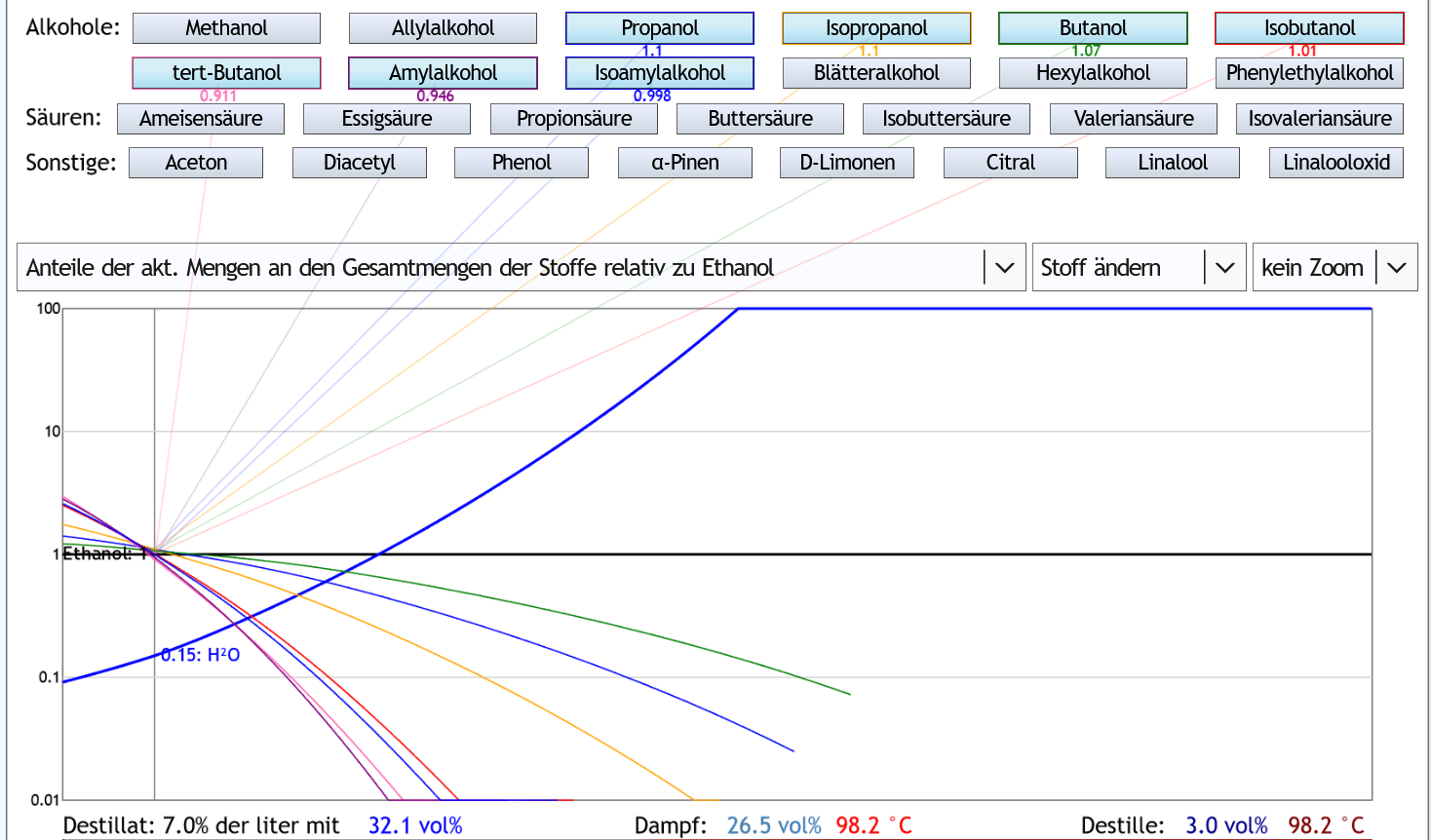 Anfangs bekommt man verglichen mit Ethanol mehr der höheren Alkohole ins Destillat, nach ca. 7% der liter weniger.
Anfangs bekommt man verglichen mit Ethanol mehr der höheren Alkohole ins Destillat, nach ca. 7% der liter weniger.
Nun 40vol%, 1 theor. Boden: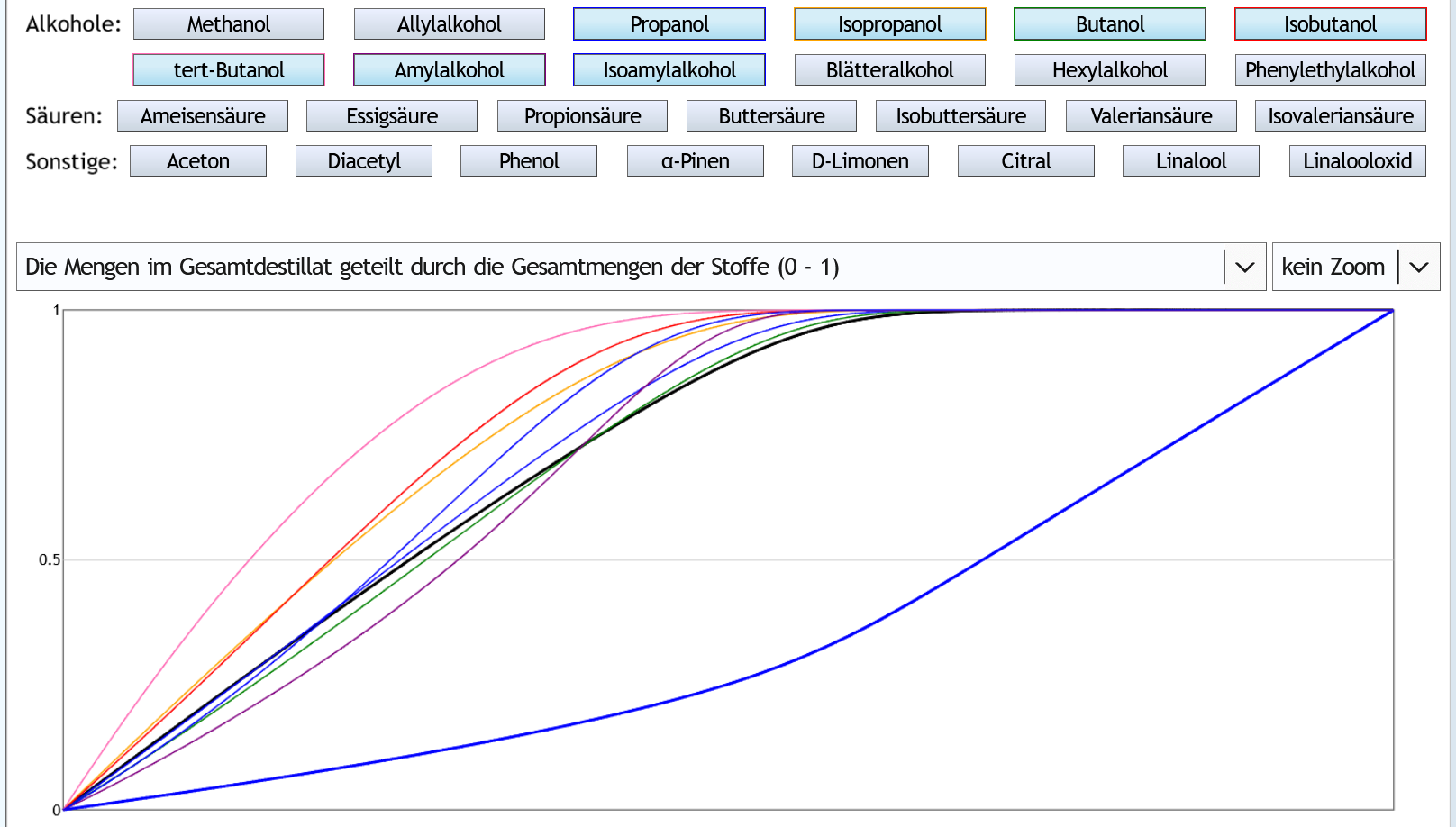 Einige Kurven sind ein wenig unterhalb der von Ethanol, andere ein wenig über Ethanol.
Es gibt also kaum Konzentrierungen oder Zurückhaltungen.
Einige Kurven sind ein wenig unterhalb der von Ethanol, andere ein wenig über Ethanol.
Es gibt also kaum Konzentrierungen oder Zurückhaltungen.
Andere Darstellung: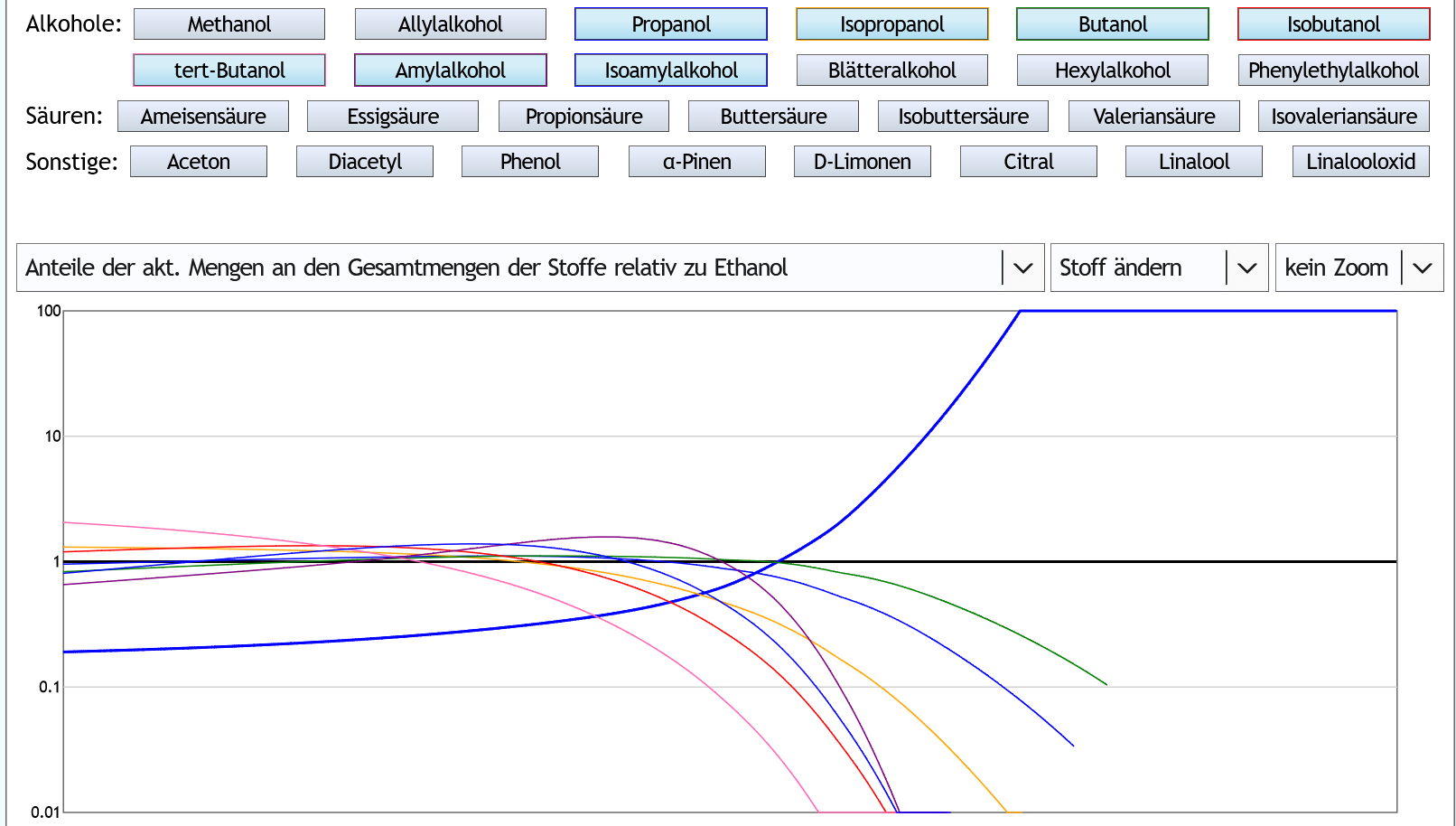 Hier sieht man dasselbe.
Der Abfall der Kurven rechts kommt daher, daß sich die höheren Alkohole insgesamt etwas schneller aufgebraucht haben als Ethanol.
Bei dem Abfall der Kurve ist die Konzentration der Stoffe in der Destille fast 0, aber Ethanol ist noch vorhanden.
Hier sieht man dasselbe.
Der Abfall der Kurven rechts kommt daher, daß sich die höheren Alkohole insgesamt etwas schneller aufgebraucht haben als Ethanol.
Bei dem Abfall der Kurve ist die Konzentration der Stoffe in der Destille fast 0, aber Ethanol ist noch vorhanden.
Nun mit Rektifikation: 5vol%, 6 theor. Böden, Zoom x10: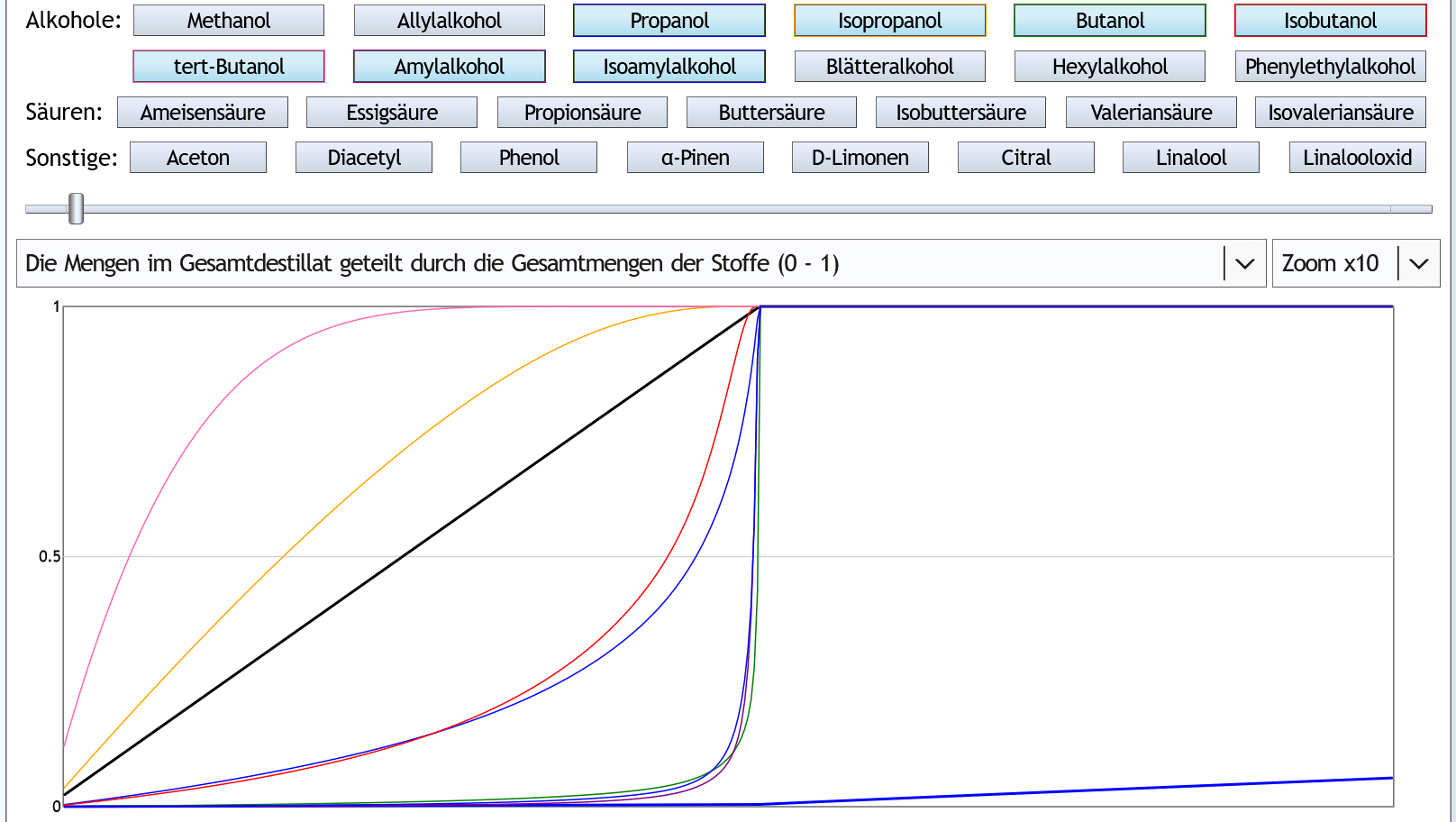 Die typischen und meisten höheren Alkohole werden im Kessel zurückgehalten und schießen, sobald der Alkoholgehalt im Kessel gesunken ist, steil nach oben.
Die Rektifikation reduziert also, daß höhere Akohole in den Vor- und Mittellauf gelangen.
Die typischen und meisten höheren Alkohole werden im Kessel zurückgehalten und schießen, sobald der Alkoholgehalt im Kessel gesunken ist, steil nach oben.
Die Rektifikation reduziert also, daß höhere Akohole in den Vor- und Mittellauf gelangen.
Andere Darstellung: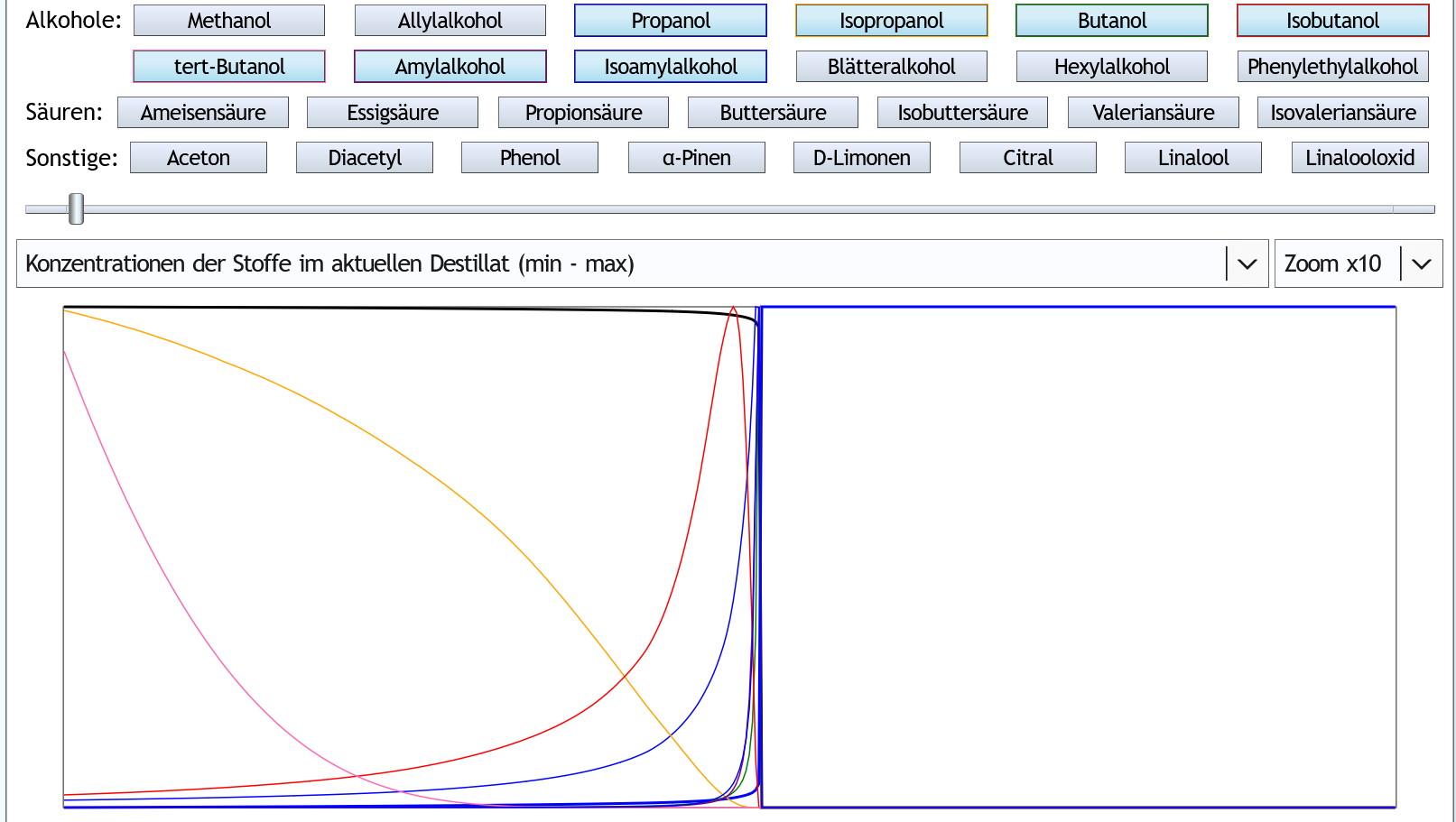 Hier sieht man, wie die aktuelle Mengen der meisten höheren Alkohole, kurz bevor der Alkoholgehalt im Dampf absackt, in die Höhe schießen.
Daß sie danach gleich wieder abfallen, liegt daran, daß sie sich sofort aufgebraucht haben, also innerhalb kürzester Zeit fast komplett im Destillat gelandet sind.
Das kennt man auch aus der Praxis:
wenn man eine starke rektifizierende Refluxdestille so lange laufen lässt, bis der Alkohol fast komplett abdestilliert ist, kommt plötzlich eine kurze Sapnne übelst riechendes Destillat.
Hier sieht man, wie die aktuelle Mengen der meisten höheren Alkohole, kurz bevor der Alkoholgehalt im Dampf absackt, in die Höhe schießen.
Daß sie danach gleich wieder abfallen, liegt daran, daß sie sich sofort aufgebraucht haben, also innerhalb kürzester Zeit fast komplett im Destillat gelandet sind.
Das kennt man auch aus der Praxis:
wenn man eine starke rektifizierende Refluxdestille so lange laufen lässt, bis der Alkohol fast komplett abdestilliert ist, kommt plötzlich eine kurze Sapnne übelst riechendes Destillat.
Nun 40vol%, 6 theor. Böden, kein Zoom: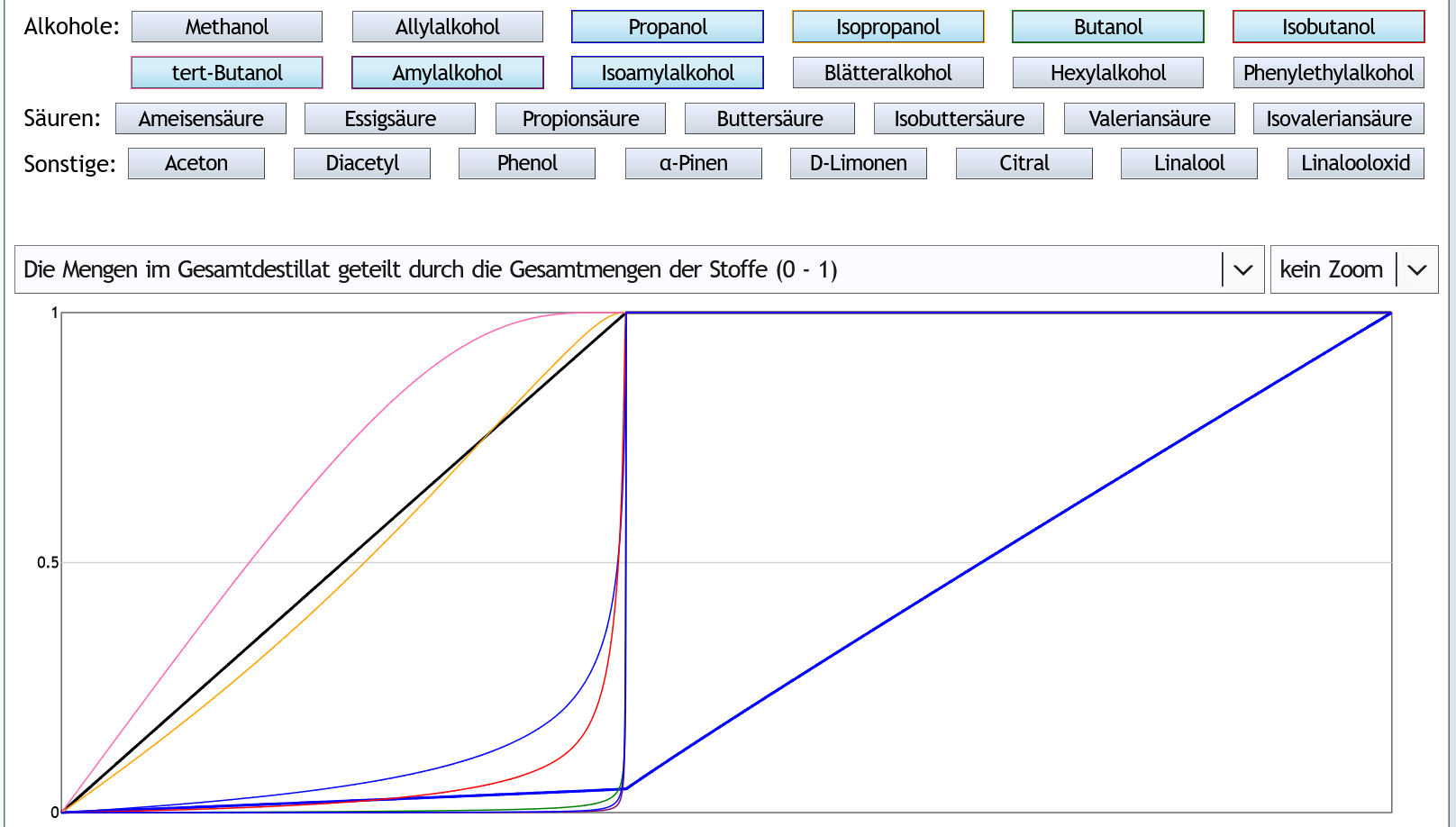
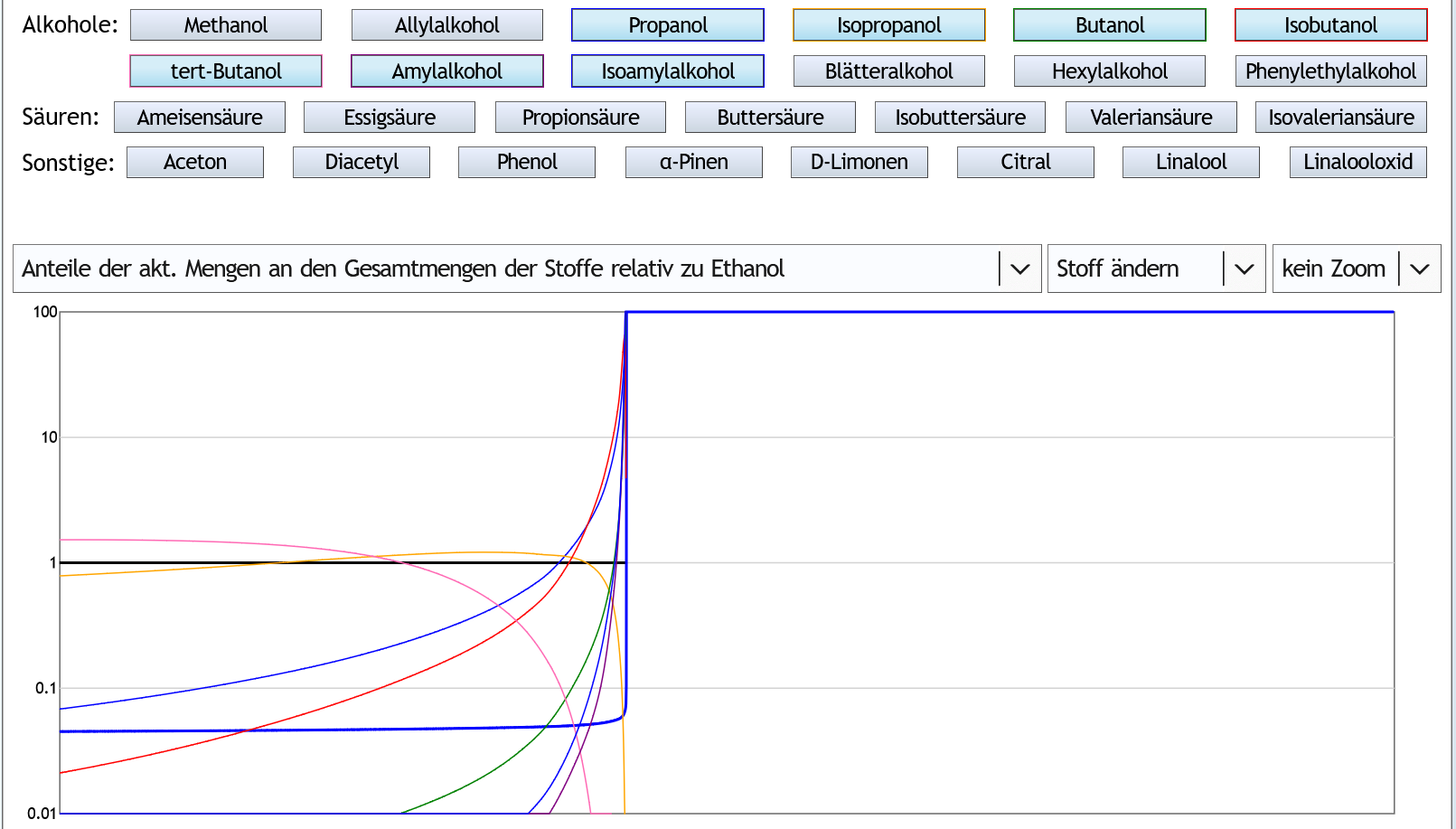 Der Zeitpunkt, an dem der Ethanolgehalt absackt, ist natürlich später.
Aber das sieht man das gleiche Verhalten, nur etwas extremer.
Der Zeitpunkt, an dem der Ethanolgehalt absackt, ist natürlich später.
Aber das sieht man das gleiche Verhalten, nur etwas extremer.
Mit höherer Rektifikation kann man die höheren Alkohole noch wesentlich stärker zurückhalten, sowohl in der Theorie als auch in der Praxis. Deren hochschießen, sobald das Ethanol destilliert ist, ist aber nicht so extrem, wie es der Simulator berechnet, da die Begleitstoffmoleküle beginnen, sich gegenseitig zu beeinflussen.
Fazit: Daß die höheren Alkohole Nachlaufstoffe sind, stimmt also nur für Refluxdestillationen.
5vol%, 1 theor. Boden:
Andere Darstellung:
Nun 40vol%, 1 theor. Boden:
Andere Darstellung:
Nun mit Rektifikation: 5vol%, 6 theor. Böden, Zoom x10:
Andere Darstellung:
Nun 40vol%, 6 theor. Böden, kein Zoom:
Mit höherer Rektifikation kann man die höheren Alkohole noch wesentlich stärker zurückhalten, sowohl in der Theorie als auch in der Praxis. Deren hochschießen, sobald das Ethanol destilliert ist, ist aber nicht so extrem, wie es der Simulator berechnet, da die Begleitstoffmoleküle beginnen, sich gegenseitig zu beeinflussen.
Fazit: Daß die höheren Alkohole Nachlaufstoffe sind, stimmt also nur für Refluxdestillationen.
Soll man auch bei Raubränden Vorlauf abtrennen?
Zuerst die Frage, was effizienter ist, also ob man beim Rau- oder beim Feinbrand den Vorlauf effizienter abtrennen kann.
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 1 berechnet.
Dargestellt werden die beiden wichtigsten Vorlaufstoffe Acetaldehyd und Ethylacetat:
5vol%, 1.3 theor. Böden, Zoom x10: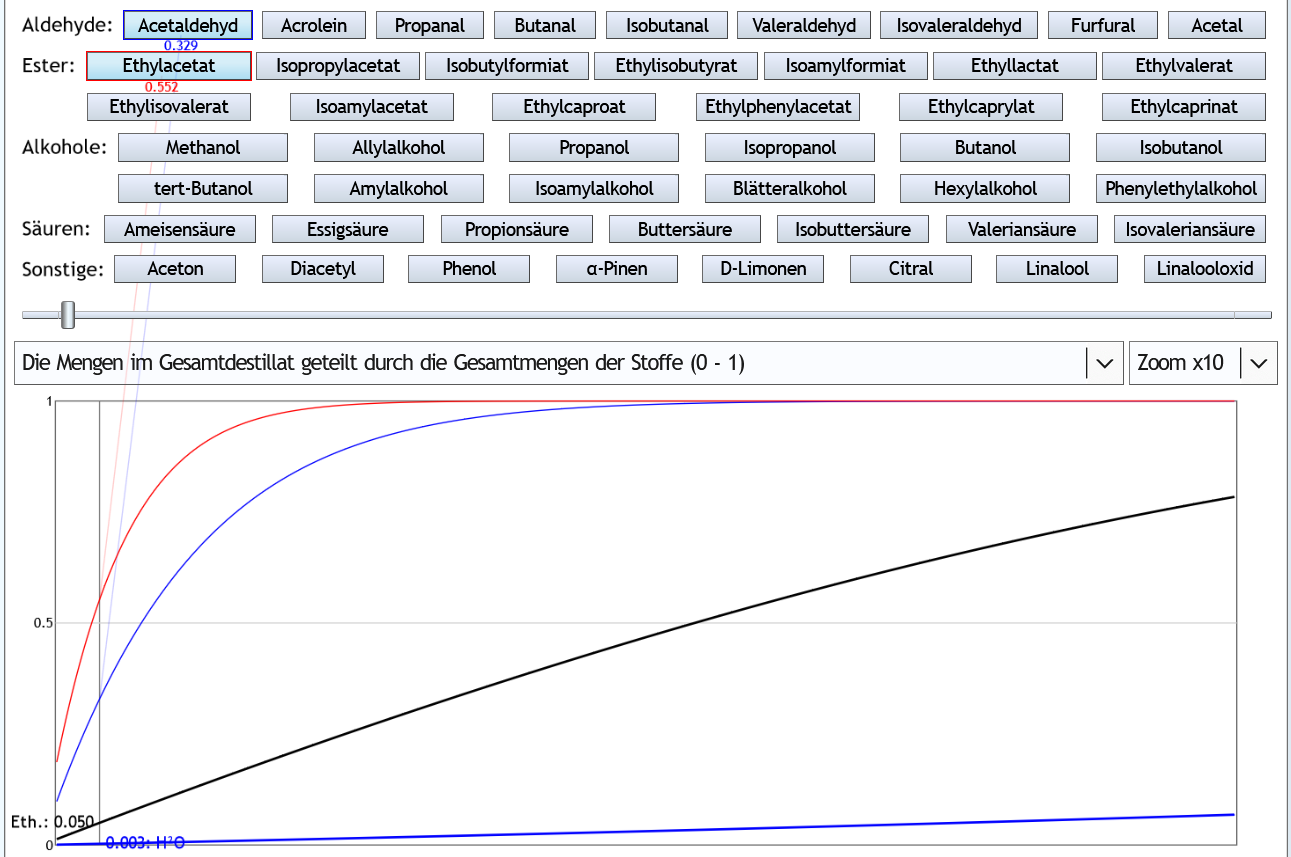 Wenn man 5% (0.050) des Ethanols als Vorlauf abtrennt, hat man 32.9% (0.329) des Acetaldehyds und 55.2% (0.552) des Ethylacetats entfernt.
Wenn man 5% (0.050) des Ethanols als Vorlauf abtrennt, hat man 32.9% (0.329) des Acetaldehyds und 55.2% (0.552) des Ethylacetats entfernt.
Nun 40vol% (Feinbrand):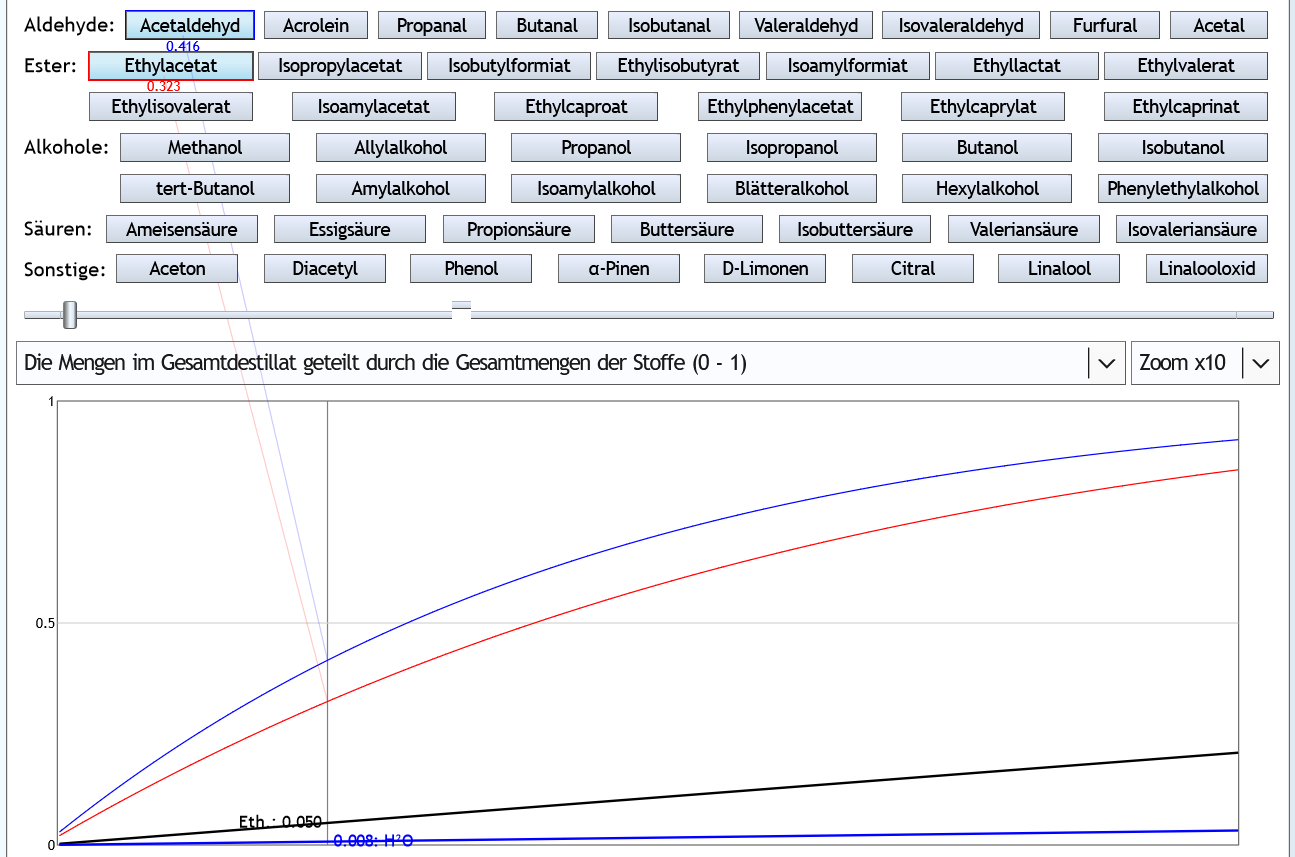 Wenn man 5% des Ethanols als Vorlauf abtrennt, hat man 41.6% des Acetaldehyds (also etwas mehr wie bei 5vol%) und 32.3% (also weniger wie bei 5%) des Ethylacetats entfernt.
Wenn man 5% des Ethanols als Vorlauf abtrennt, hat man 41.6% des Acetaldehyds (also etwas mehr wie bei 5vol%) und 32.3% (also weniger wie bei 5%) des Ethylacetats entfernt.
Das Entfernen des Vorlaufs ist also beim Raubrand insgesamt etwas effizienter als beim Feinbrand.
Zwei Punkte relativieren dieses Ergebnis allerdings:
- Nach dem Raubrand kann neuer Vorlauf entstehen. Also ein Abtrennen beim Feinbrand muss meist trotzdem gemacht werden.
- Wird der Feinbrand mit einer Refluxdestille gemacht, ist das Abtrennen natürlich wesentlich effizienter: 40vol%, 6 theor. Böden: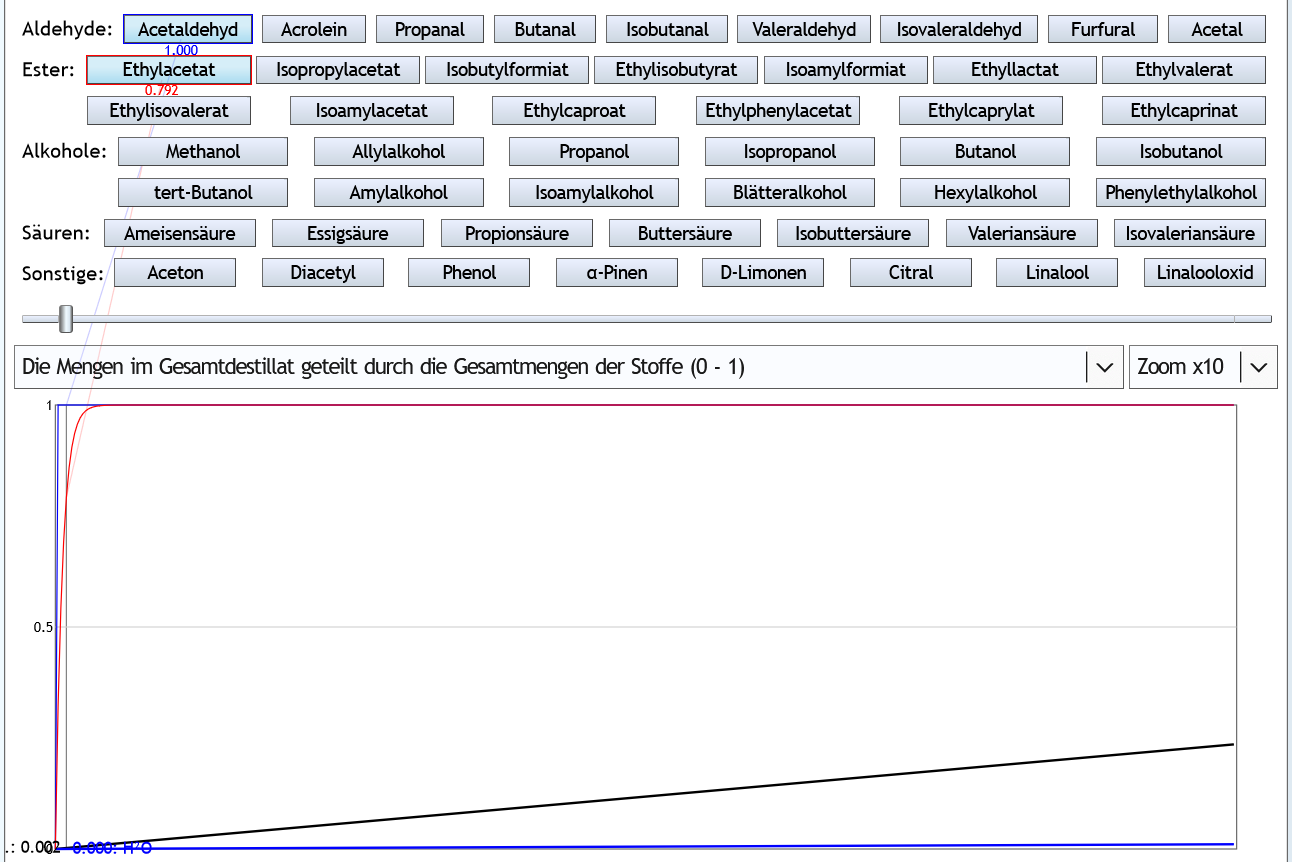 Schon nach 2% des Ethanols sind beide Vorlaufstoffe recht vollständig entfernt.
Ob das wirklich ganz so komplett und schnell auch in der Praxis passiert, ist allerdings nicht sicher.
Aber am allgemeinen Ergebnis ändert das nichts.
Schon nach 2% des Ethanols sind beide Vorlaufstoffe recht vollständig entfernt.
Ob das wirklich ganz so komplett und schnell auch in der Praxis passiert, ist allerdings nicht sicher.
Aber am allgemeinen Ergebnis ändert das nichts.
Meist geht es aber nicht darum, möglichst wenig Ethanol beim Vorlaufabtrennen zu verlieren, sondern man möchte das geschmacklich beste Ergebnis haben. Man möchte beim Vorlaufabtrennen möglichst viele schlechte Aromen abtrennen, aber möglichst ohne, daß dabei auch viele gute Aromen abgetrennt werden.
Und bei dieser Frage kann eine Simulation durch seine vergleichende Darstellungen Antworten liefern, indem sie darstellt, wie sich innerhalb einer Stoffgruppe die eher guten und die eher schlechten Aromastoffe zueinander verhalten. Die eher schlechten Stoffe sind die mit den niedrigen Molmassen und die eher guten die mit hohen Molmassen. Und nach Molmasse sind die Stoffe im ja Simulator geordnet. Also das schlechteste (und gleichzeitig häufigste) Aldehyd ist Acetalehyd.
5vol%, 1.3 theor. Böden, Zoom x10 (nur von der Molekularstruktur bzw. Summenformel typische Aldehyde bzw. mit Acetaldehyd vergleichbare Aldehyde sind dargestellt):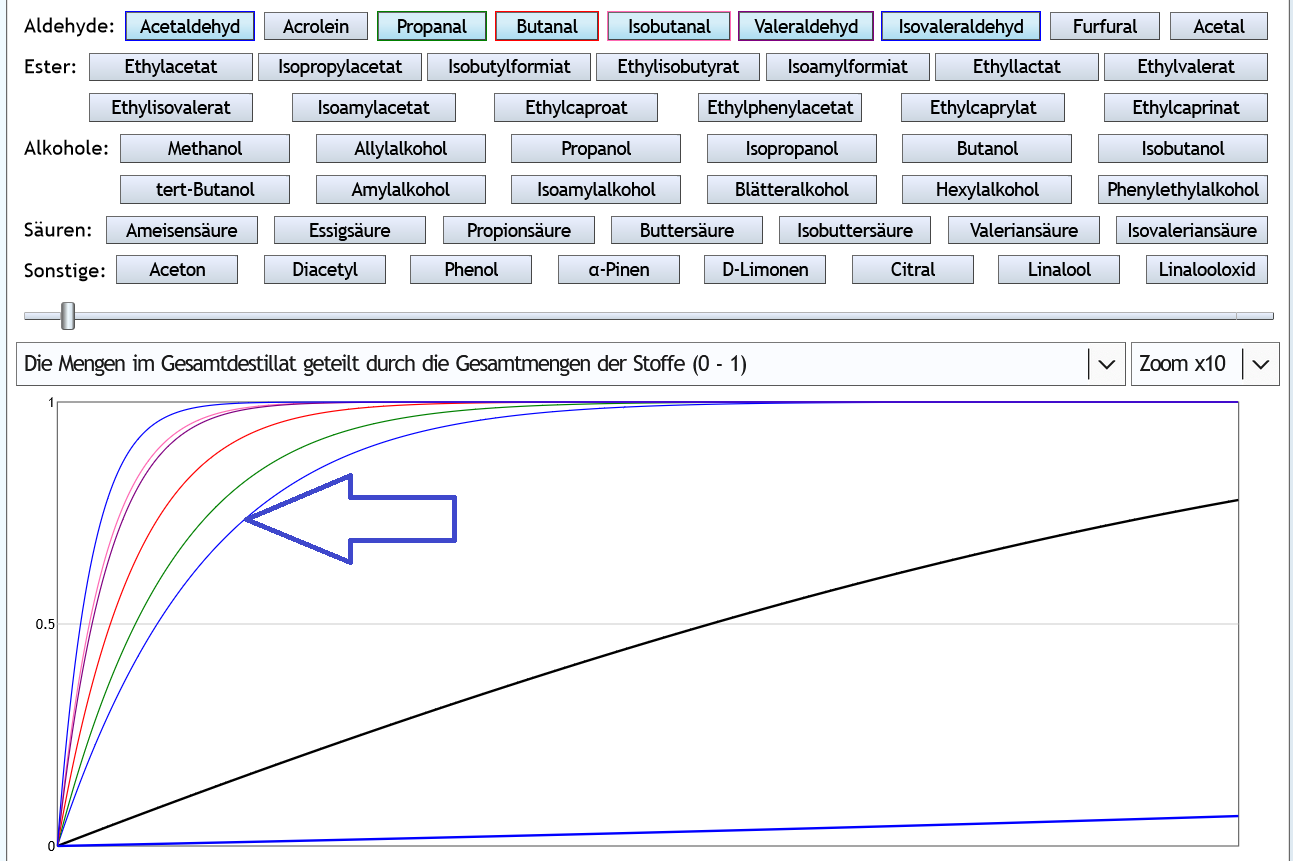 Acetaldehyd (dünne blaue Linie, siehe Pfeil) konzentriert sich am wenigsten.
Beim Vorlaufabtrennen reduziert man also in diesem Fall mehr die guten als die schlechten Aromen.
Acetaldehyd (dünne blaue Linie, siehe Pfeil) konzentriert sich am wenigsten.
Beim Vorlaufabtrennen reduziert man also in diesem Fall mehr die guten als die schlechten Aromen.
Andere Darstellung: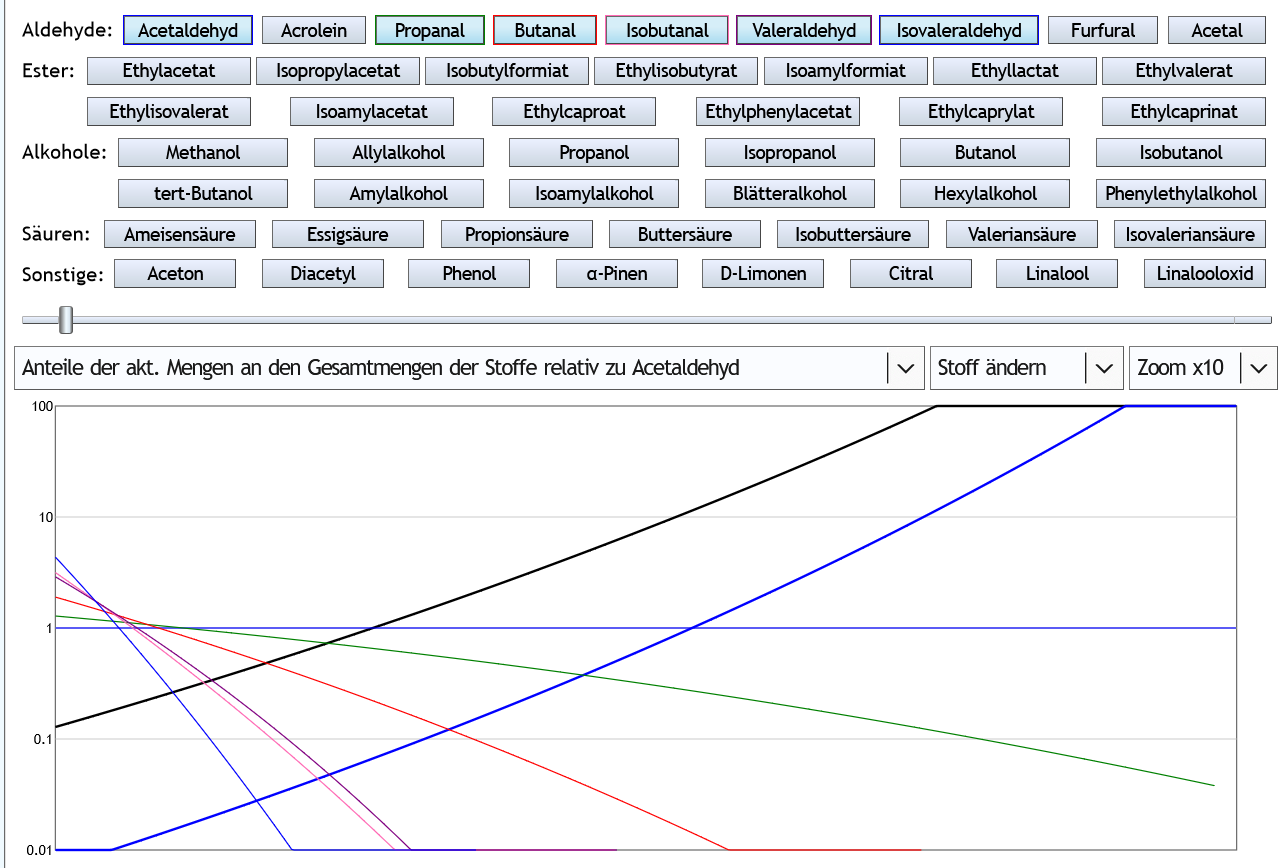 Alle Kurven beginnen über der 1, also über der Kurve von Acetaldehyd.
Alle Kurven beginnen über der 1, also über der Kurve von Acetaldehyd.
Nun 40vol% (also Feinbrandstärke):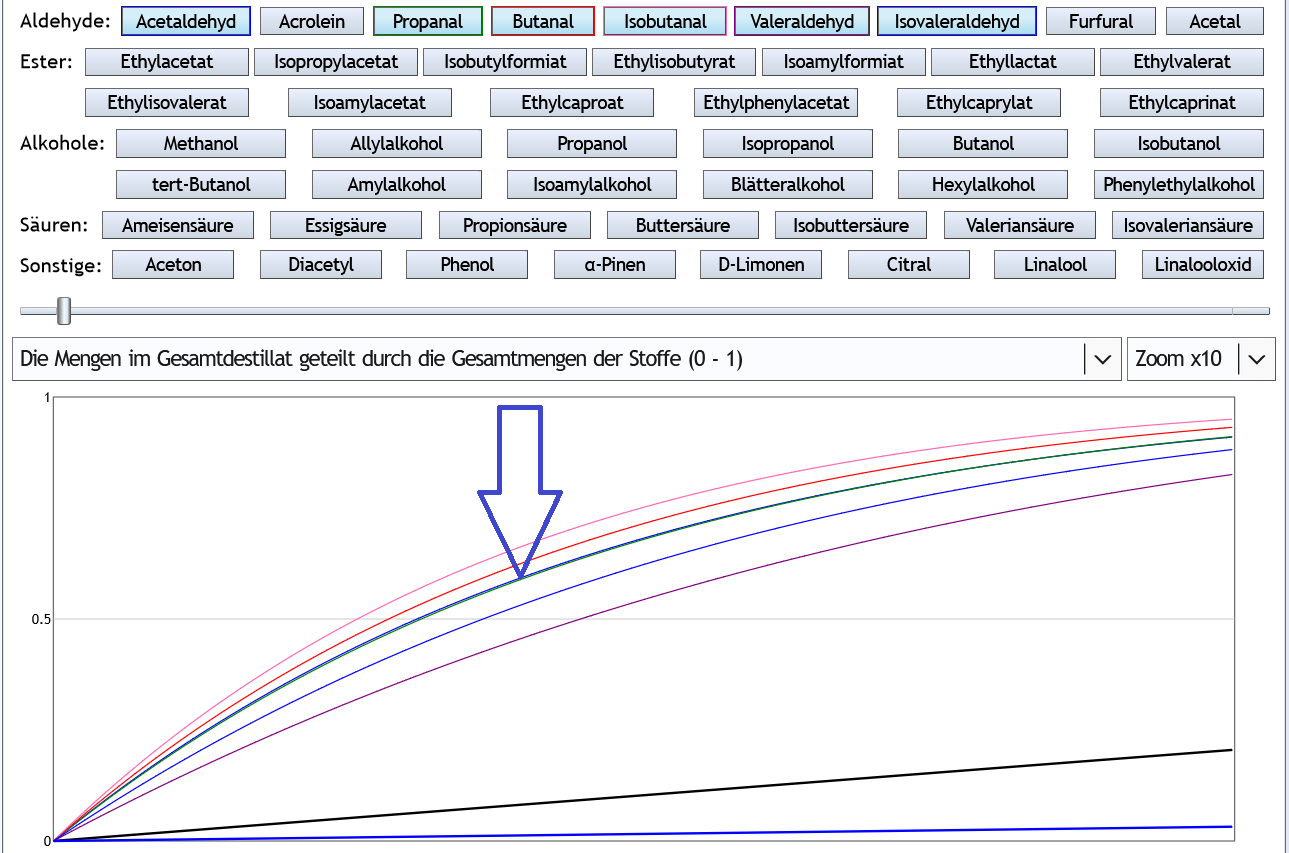 Bei höherem Alkoholgehalt konzentriert sich Acetaldehyd (Pfeil) besser als die wertvolleren Valeraldehyd und Isovaleraldehyd.
Bei höherem Alkoholgehalt konzentriert sich Acetaldehyd (Pfeil) besser als die wertvolleren Valeraldehyd und Isovaleraldehyd.
Andere Ansicht: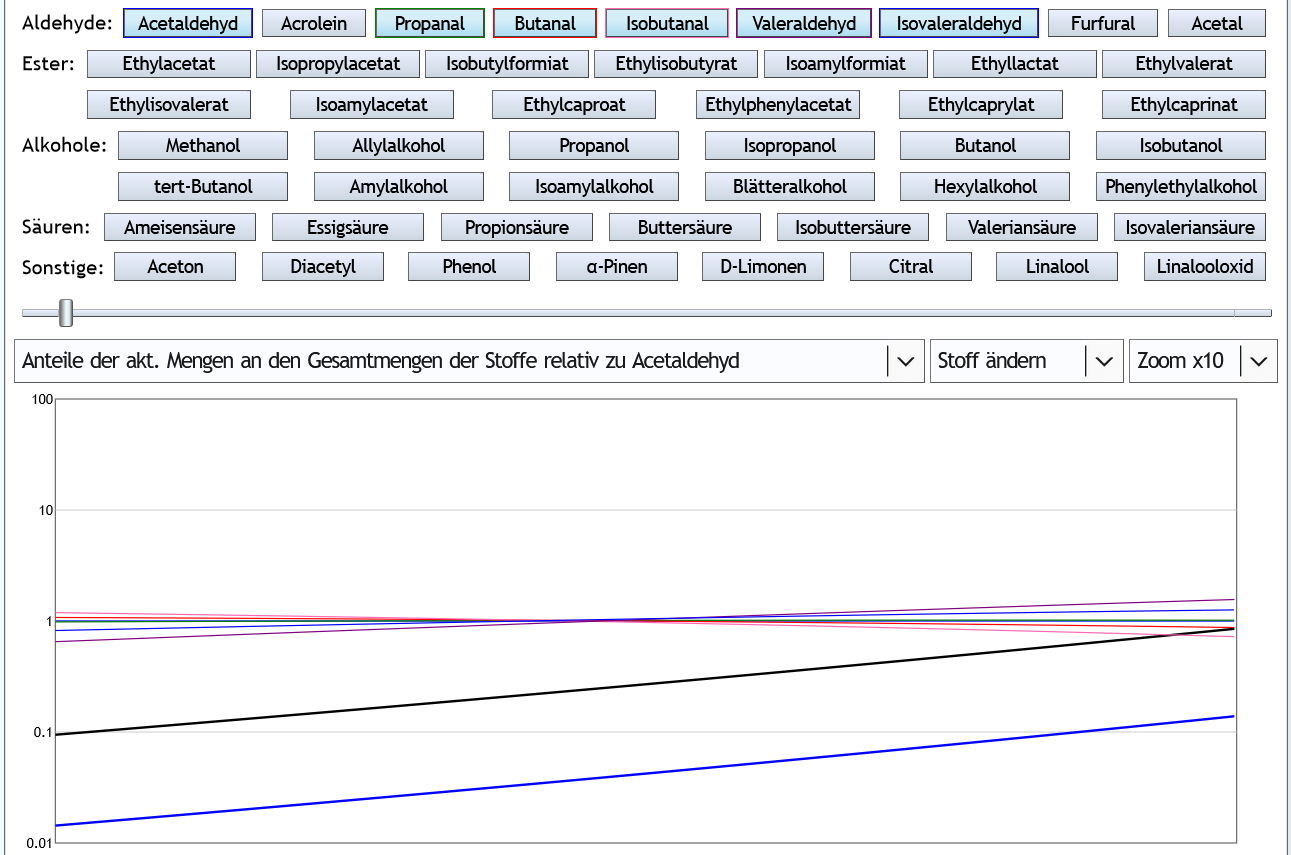 Valeraldehyd und Isovaleraldehyd sind am Anfang unter 1.
Valeraldehyd und Isovaleraldehyd sind am Anfang unter 1.
Fazit: Bezüglich der Aldehyde ist ein Vorlaufabtrennen beim Raubrand also eher aromaschädlich.
Nun die Ester:
5vol%, 1.3 theor. Böden, Zoom x10 (nur von der Molekularstruktur bzw. Summenformel typische Ester bzw. mit Ethylacetat vergleichbare Ester sind dargestellt):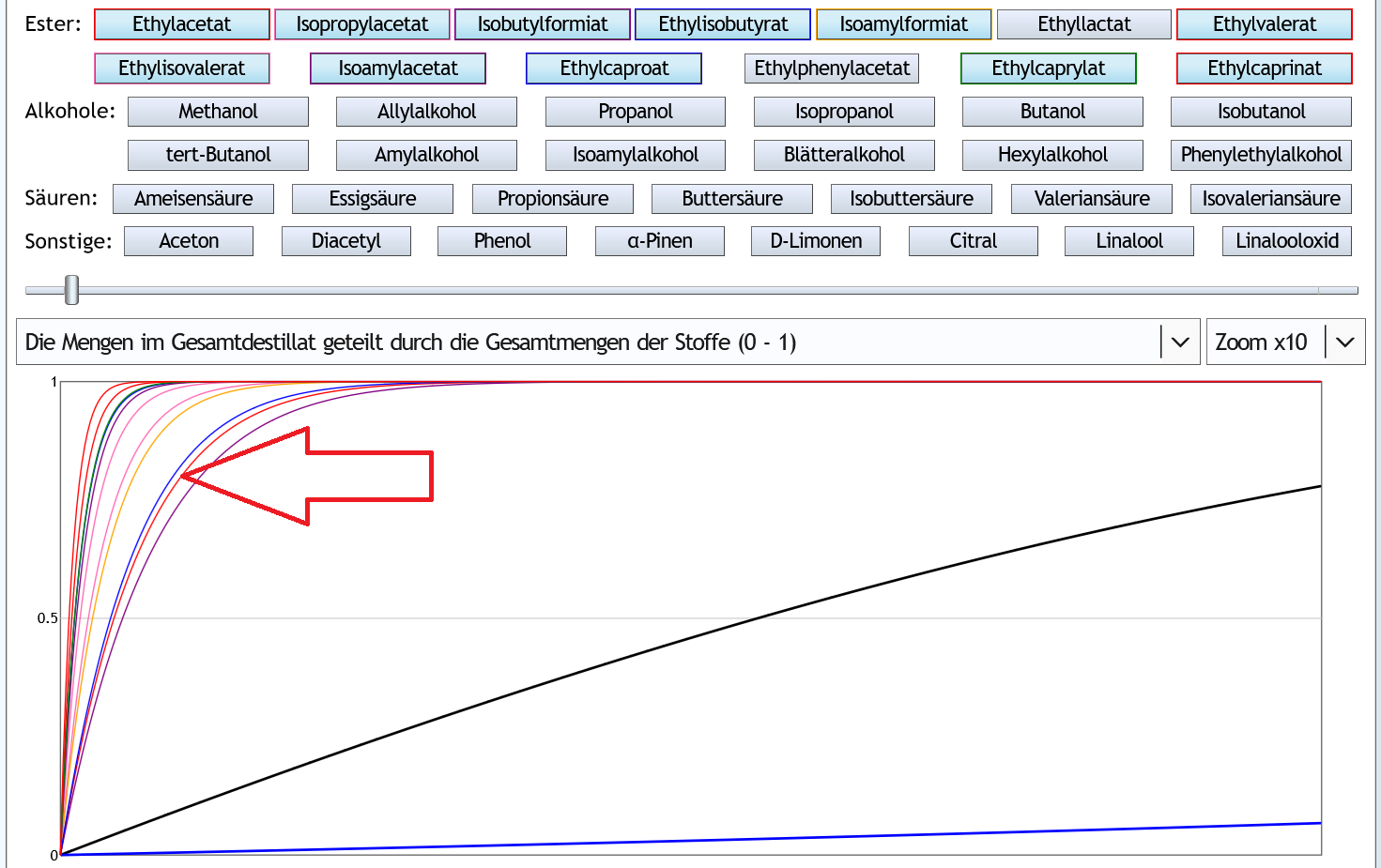 Fast alle Stoffe konzentieren sich mehr im Vorlauf als Ethylacetat (dünne rote Linie, siehe Pfeil).
Beim Vorlaufabtrennen reduziert man in diesem Fall also mehr die guten als die schlechten Aromen.
Fast alle Stoffe konzentieren sich mehr im Vorlauf als Ethylacetat (dünne rote Linie, siehe Pfeil).
Beim Vorlaufabtrennen reduziert man in diesem Fall also mehr die guten als die schlechten Aromen.
Andere Darstellung: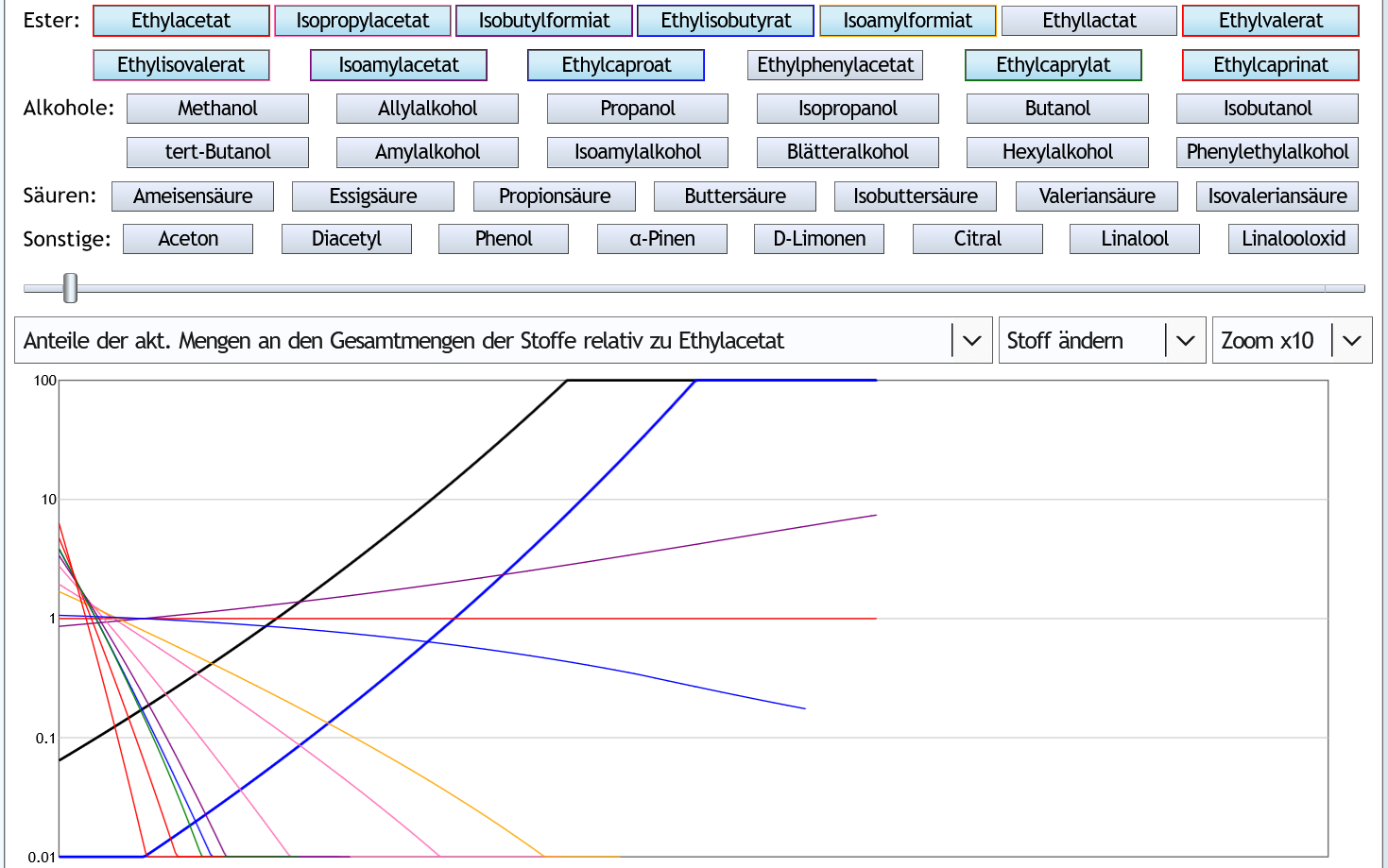 Alle Kurven beginnen über der 1, also über der Kurve von Ethylacetat.
Alle Kurven beginnen über der 1, also über der Kurve von Ethylacetat.
Nun 40vol% (also Feinbrandstärke):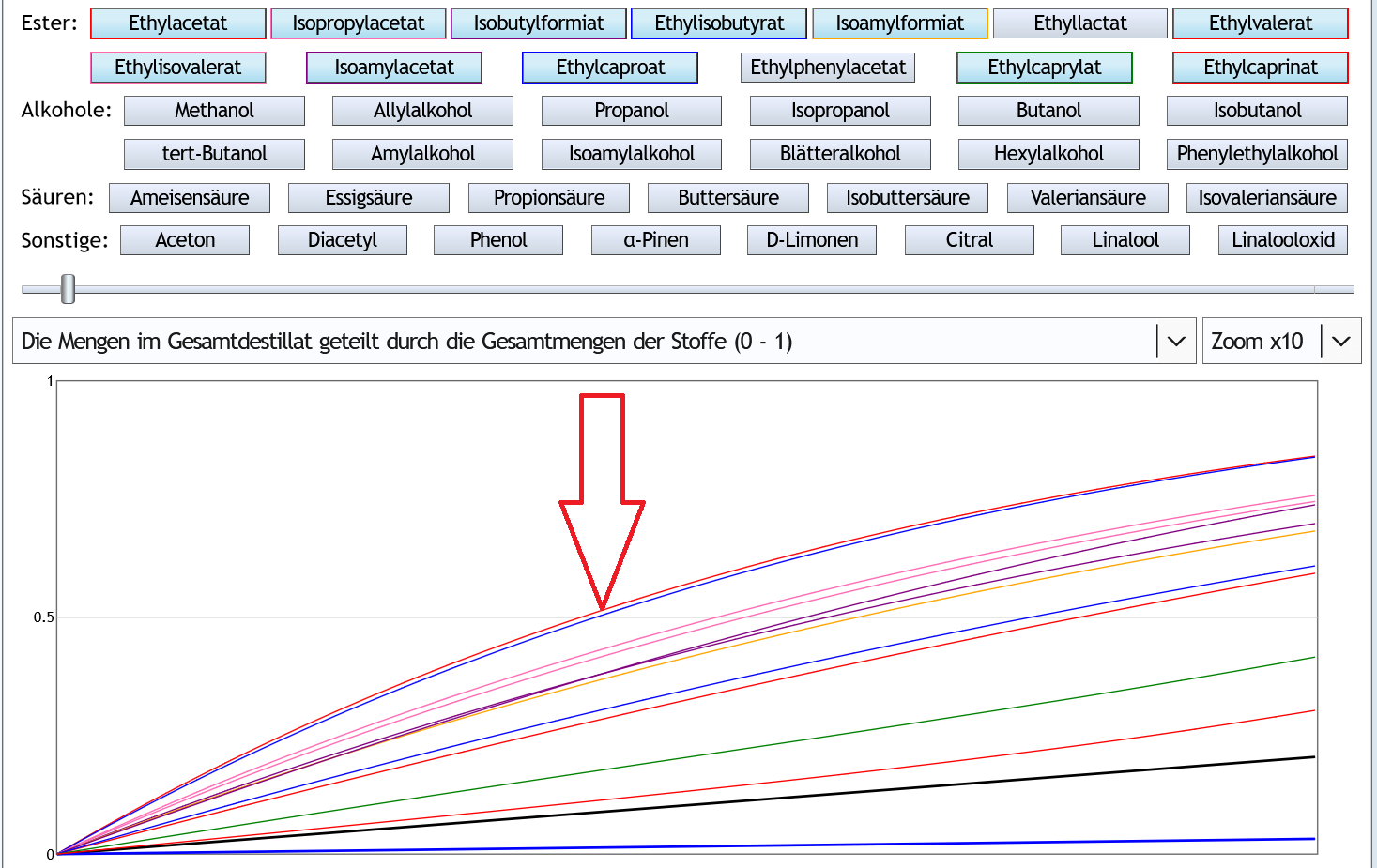 Bei höherem Alkoholgehalt konzentriert sich Ethylacetat (dünne rote Linie, siehe Pfeil) im Vorlauf besser als alle anderen (wertvolleren) Ester.
Bei höherem Alkoholgehalt konzentriert sich Ethylacetat (dünne rote Linie, siehe Pfeil) im Vorlauf besser als alle anderen (wertvolleren) Ester.
Andere Darstellung: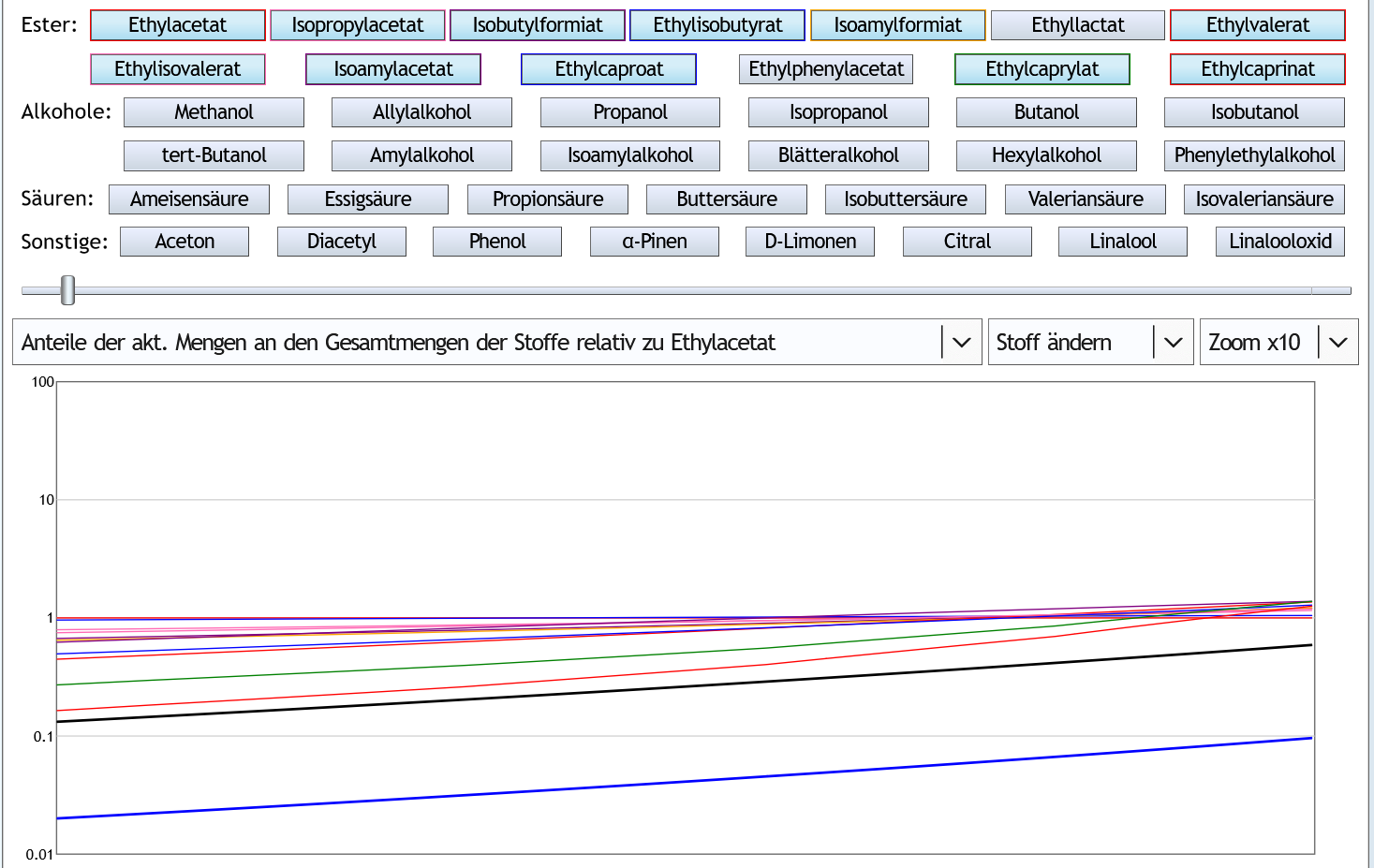 Alle Kurven beginnen unter der 1.
Alle Kurven beginnen unter der 1.
Fazit: Bezüglich der Ester ist ein Vorlaufabtrennen beim Raubrand also ebenfalls aromaschädigend.
Mit dem Begleitstoffesimulator 2 kann man sich das noch etwas anders anschauen:
10lt mit 8vol%.
A)
Raubrand, Vorlauf bis 0.01lt Destillat mit 1.5 theor. Böden, dann bis 2.49lt Destillat mit 1.1 theor. Böden, Vorlauf nicht weggeschüttet.
Feinbrand, Vorlauf bis 0.02lt Destillat mit 1.5 theor. Böden, dann Mittellauf bis aktuell 63vol% mit 1.2 theor. Böden. Die Daten des Mittellaufs: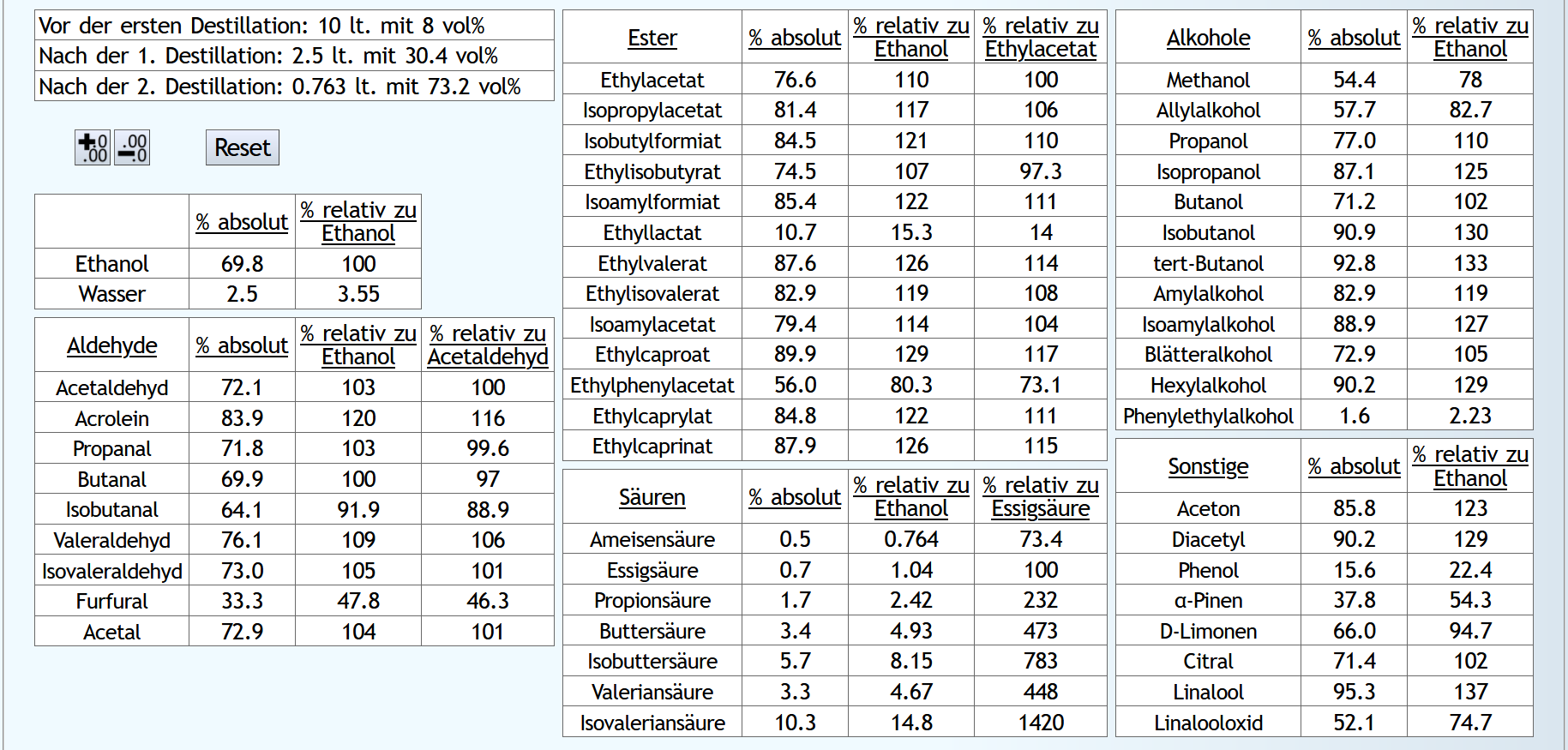 B)
B)
Raubrand, Vorlauf bis 0.01lt Destillat mit 1.5 theor. Böden, dann bis 2.49lt Destillat mit 1.1 theor. Böden, Vorlauf weggeschüttet.
Feinbrand, Vorlauf bis 0.01lt Destillat mit 1.5 theor. Böden, dann Mittellauf bis aktuell 63vol% mit 1.2 theor. Böden. Die Daten des Mittellaufs: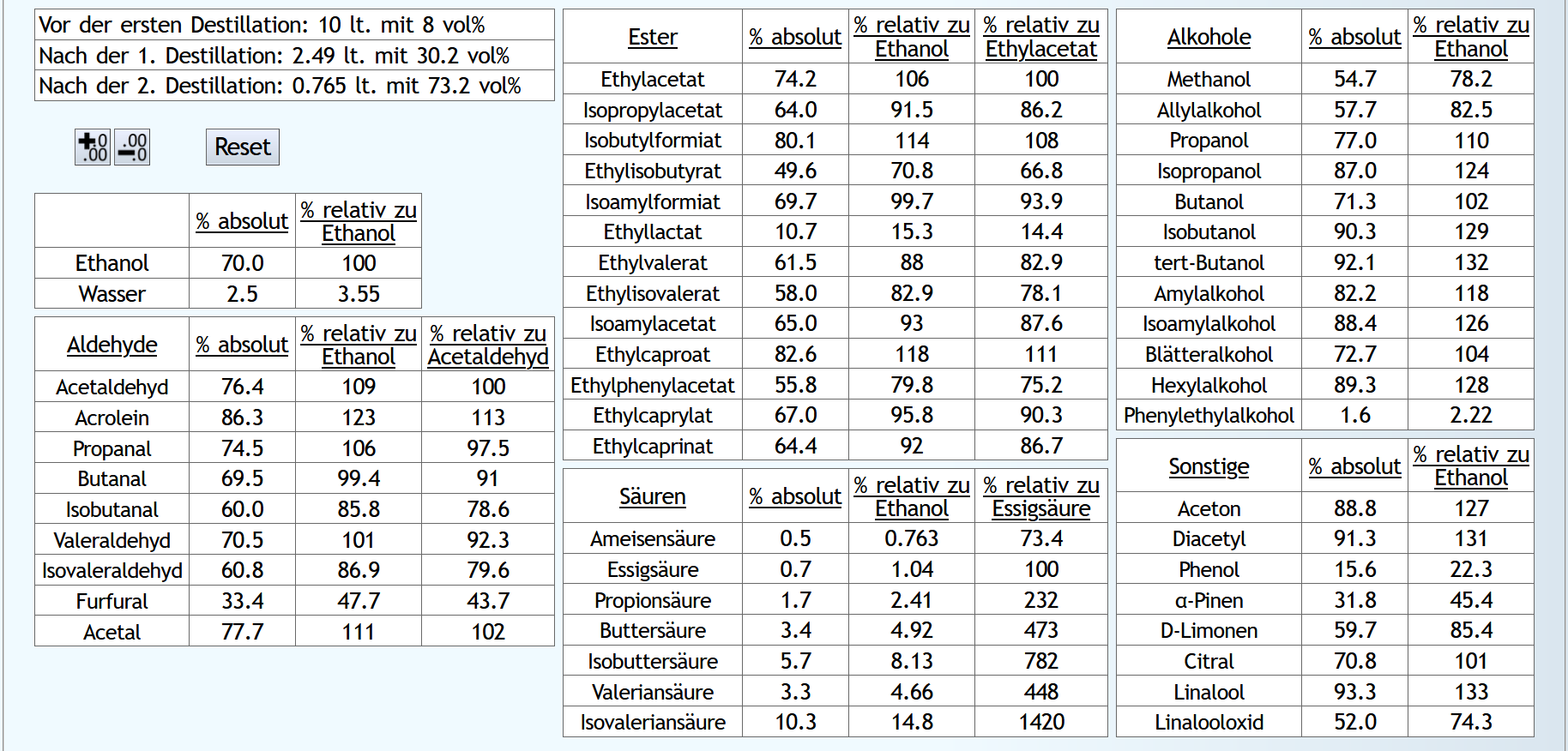 B ist allgemein etwas neutraler.
Weniger Begleitstoffe verglichen mit Ethanol.
Bei den Aldehyden nicht unbedingt, aber bei den Estern.
B ist allgemein etwas neutraler.
Weniger Begleitstoffe verglichen mit Ethanol.
Bei den Aldehyden nicht unbedingt, aber bei den Estern.
B ist qualitativ etwas schlechter, minimal bei den größeren Aldehyden (Valeraldehyd, Isovaleraldehyd) verglichen mit Acetaldehyd, und bei fast allen Estern im Vergleich zu Ethylacetat. Und bei einigen Terpenen (Sonstige) macht es auch was aus.
Dieser sehr kleine Unterschied des Destillationsprozesses macht aber natürlich nicht viel aus. Aber generell sieht man schon, daß ein Abtrennen des Vorlaufs teilweise beim Raubrand erstens einen etwas sauberen Brand erzeugt, zweitens aber eher die guten als die schlechten Aromastoffe abgetrennt werden.
Fazit: Wenn man Neutralalkohol brennt, sollte man jeden Brand für die Vorlaufabtrennung nutzen. Denn die unterschiedliche Alkoholstärke jedesmal hilft: manche Stoffe lassen sich eher bei niedriger Alkoholstärke effizient abtrennen und manche eher bei hoher Alkoholstärke. Bei Aromabränden sollte man aber nur beim letzten Brand, also bei dem mit der höchsten Alkoholstärke, Vorlauf abtrennen, da eine hohe Alkoholstärke bewirkt, daß mehr schlechte als gute Stoffe im Vorlauf landen.
Dargestellt werden die beiden wichtigsten Vorlaufstoffe Acetaldehyd und Ethylacetat:
5vol%, 1.3 theor. Böden, Zoom x10:
Nun 40vol% (Feinbrand):
Das Entfernen des Vorlaufs ist also beim Raubrand insgesamt etwas effizienter als beim Feinbrand.
Zwei Punkte relativieren dieses Ergebnis allerdings:
- Nach dem Raubrand kann neuer Vorlauf entstehen. Also ein Abtrennen beim Feinbrand muss meist trotzdem gemacht werden.
- Wird der Feinbrand mit einer Refluxdestille gemacht, ist das Abtrennen natürlich wesentlich effizienter: 40vol%, 6 theor. Böden:
Meist geht es aber nicht darum, möglichst wenig Ethanol beim Vorlaufabtrennen zu verlieren, sondern man möchte das geschmacklich beste Ergebnis haben. Man möchte beim Vorlaufabtrennen möglichst viele schlechte Aromen abtrennen, aber möglichst ohne, daß dabei auch viele gute Aromen abgetrennt werden.
Und bei dieser Frage kann eine Simulation durch seine vergleichende Darstellungen Antworten liefern, indem sie darstellt, wie sich innerhalb einer Stoffgruppe die eher guten und die eher schlechten Aromastoffe zueinander verhalten. Die eher schlechten Stoffe sind die mit den niedrigen Molmassen und die eher guten die mit hohen Molmassen. Und nach Molmasse sind die Stoffe im ja Simulator geordnet. Also das schlechteste (und gleichzeitig häufigste) Aldehyd ist Acetalehyd.
5vol%, 1.3 theor. Böden, Zoom x10 (nur von der Molekularstruktur bzw. Summenformel typische Aldehyde bzw. mit Acetaldehyd vergleichbare Aldehyde sind dargestellt):
Andere Darstellung:
Nun 40vol% (also Feinbrandstärke):
Andere Ansicht:
Fazit: Bezüglich der Aldehyde ist ein Vorlaufabtrennen beim Raubrand also eher aromaschädlich.
Nun die Ester:
5vol%, 1.3 theor. Böden, Zoom x10 (nur von der Molekularstruktur bzw. Summenformel typische Ester bzw. mit Ethylacetat vergleichbare Ester sind dargestellt):
Andere Darstellung:
Nun 40vol% (also Feinbrandstärke):
Andere Darstellung:
Fazit: Bezüglich der Ester ist ein Vorlaufabtrennen beim Raubrand also ebenfalls aromaschädigend.
Mit dem Begleitstoffesimulator 2 kann man sich das noch etwas anders anschauen:
10lt mit 8vol%.
A)
Raubrand, Vorlauf bis 0.01lt Destillat mit 1.5 theor. Böden, dann bis 2.49lt Destillat mit 1.1 theor. Böden, Vorlauf nicht weggeschüttet.
Feinbrand, Vorlauf bis 0.02lt Destillat mit 1.5 theor. Böden, dann Mittellauf bis aktuell 63vol% mit 1.2 theor. Böden. Die Daten des Mittellaufs:
Raubrand, Vorlauf bis 0.01lt Destillat mit 1.5 theor. Böden, dann bis 2.49lt Destillat mit 1.1 theor. Böden, Vorlauf weggeschüttet.
Feinbrand, Vorlauf bis 0.01lt Destillat mit 1.5 theor. Böden, dann Mittellauf bis aktuell 63vol% mit 1.2 theor. Böden. Die Daten des Mittellaufs:
B ist qualitativ etwas schlechter, minimal bei den größeren Aldehyden (Valeraldehyd, Isovaleraldehyd) verglichen mit Acetaldehyd, und bei fast allen Estern im Vergleich zu Ethylacetat. Und bei einigen Terpenen (Sonstige) macht es auch was aus.
Dieser sehr kleine Unterschied des Destillationsprozesses macht aber natürlich nicht viel aus. Aber generell sieht man schon, daß ein Abtrennen des Vorlaufs teilweise beim Raubrand erstens einen etwas sauberen Brand erzeugt, zweitens aber eher die guten als die schlechten Aromastoffe abgetrennt werden.
Fazit: Wenn man Neutralalkohol brennt, sollte man jeden Brand für die Vorlaufabtrennung nutzen. Denn die unterschiedliche Alkoholstärke jedesmal hilft: manche Stoffe lassen sich eher bei niedriger Alkoholstärke effizient abtrennen und manche eher bei hoher Alkoholstärke. Bei Aromabränden sollte man aber nur beim letzten Brand, also bei dem mit der höchsten Alkoholstärke, Vorlauf abtrennen, da eine hohe Alkoholstärke bewirkt, daß mehr schlechte als gute Stoffe im Vorlauf landen.
Soll man den Vorlauf von Aromabränden mit oder ohne Rektifikation brennen?
Daß das mit Rektifikation effektiver ist, also daß man so weniger Ethanol verliert, steht außer Frage.
Aber kann es vielleicht sein, daß man auf diese Weise mehr von den schlechten Vorlaufstoffen verliert und weniger von den guten?
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 1 berechnet.
40vol%, 1 theor. Boden, Zoom x10: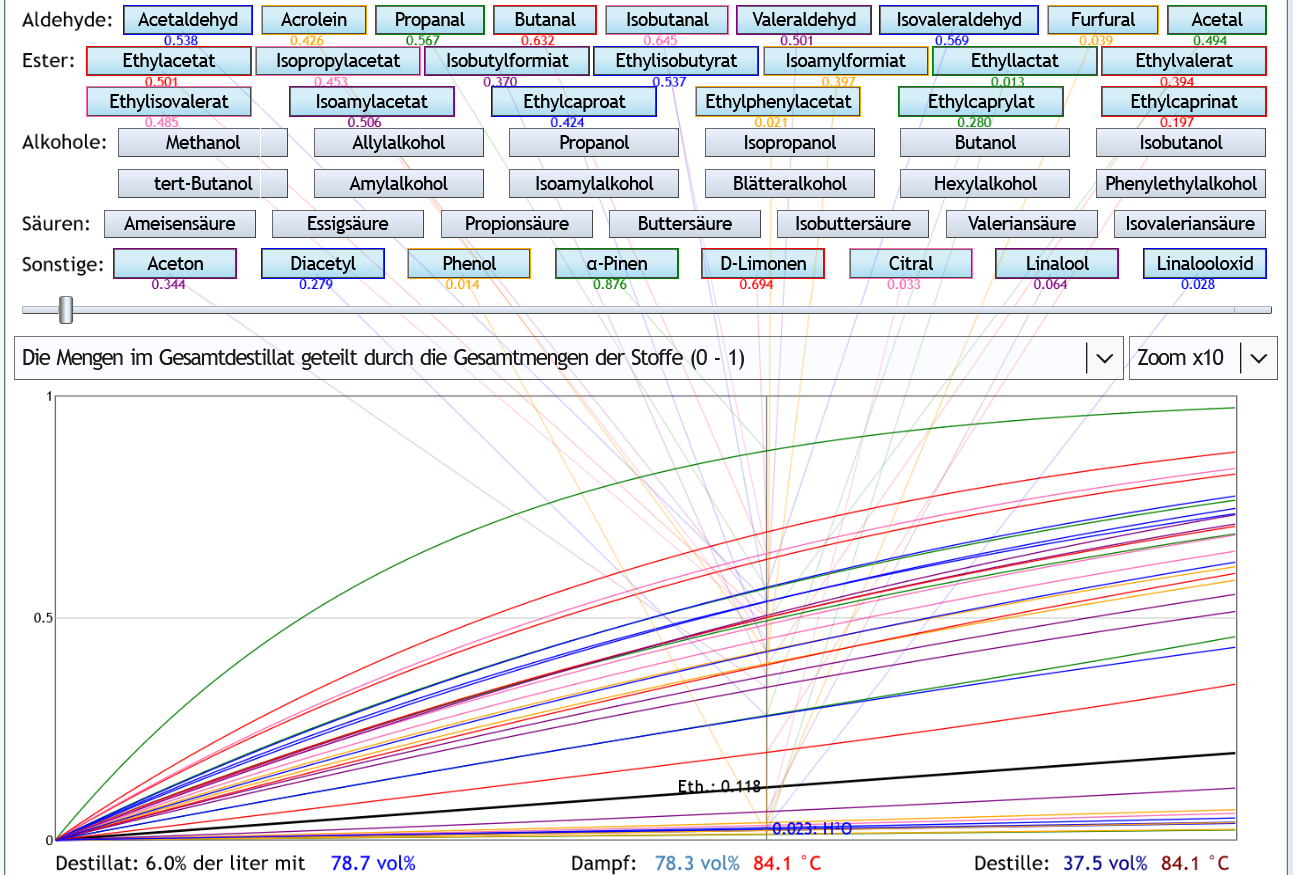 Die Werte unter den Stoffen sind an dem Punkt, wo mindestens 50% (0.5) sowohl von Acetaldehyd als auch Ethylacetat abgetrennt sind.
Man sieht, wie auch von wertvollen Stoffen schon viel in den Vorlauf gekommen ist.
Die Werte unter den Stoffen sind an dem Punkt, wo mindestens 50% (0.5) sowohl von Acetaldehyd als auch Ethylacetat abgetrennt sind.
Man sieht, wie auch von wertvollen Stoffen schon viel in den Vorlauf gekommen ist.
Nun mit 6 theor. Böden: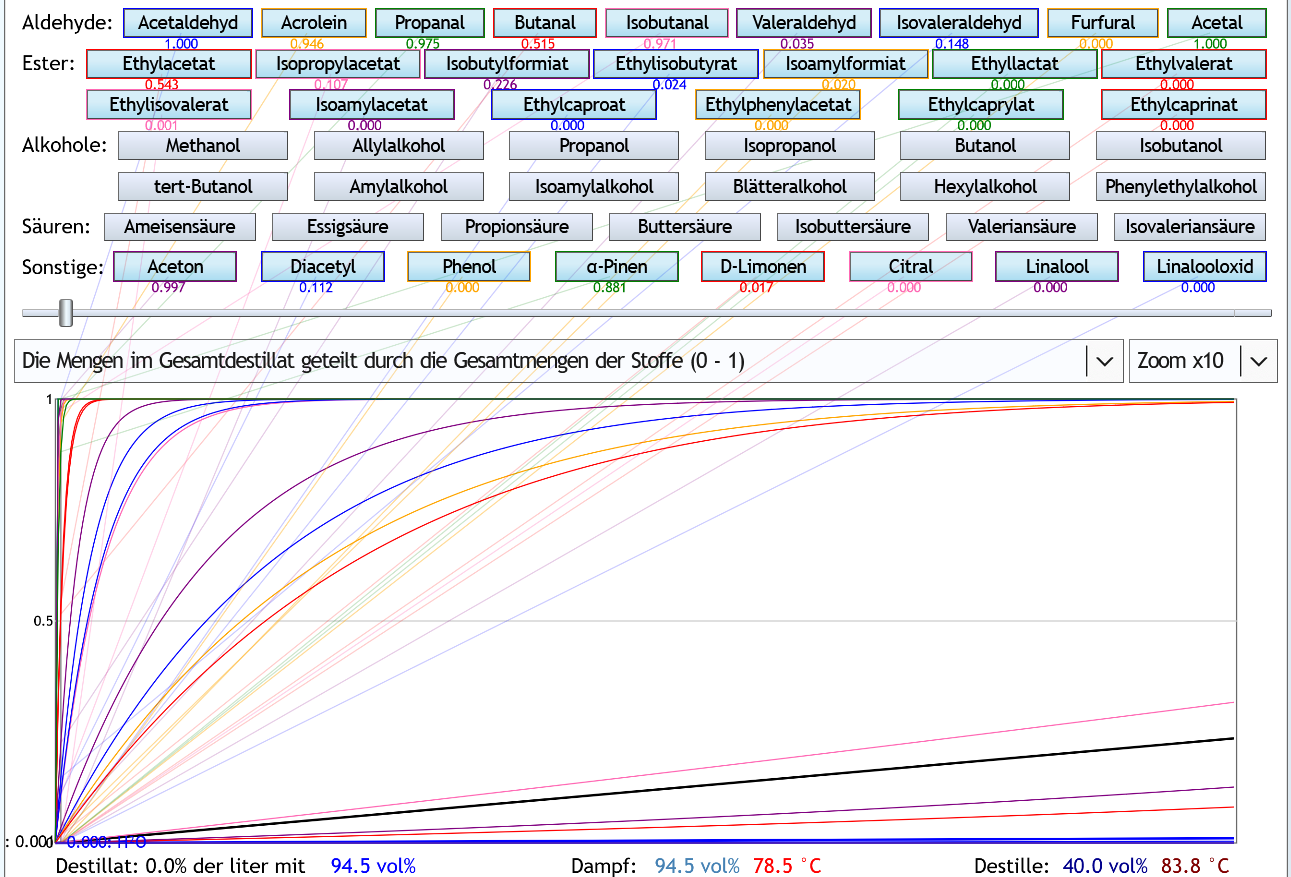 Die Werte sind da, wo ebenfalls mindestens 50% an Ethylacetat entfernt wurde wie ohne Rektifikation.
Acetaldehyd ist da bereits zu 100% abgetrennt.
Schlechte Stoffe wie Acrolein oder Aceton werden nun ebenfalls viel effektiver mit dem Vorlauf abgetrennt.
Wertvolle Stoffe wie die großen Aldehyde, die großen Ester und die Zitrusketone und -terpene (und damit wahrscheinlich auch andere wertvolle Ketone und Terpene, von denen wir leider keine Daten haben) in der untersten Reihe rechts werden geschohnt.
Die Werte sind da, wo ebenfalls mindestens 50% an Ethylacetat entfernt wurde wie ohne Rektifikation.
Acetaldehyd ist da bereits zu 100% abgetrennt.
Schlechte Stoffe wie Acrolein oder Aceton werden nun ebenfalls viel effektiver mit dem Vorlauf abgetrennt.
Wertvolle Stoffe wie die großen Aldehyde, die großen Ester und die Zitrusketone und -terpene (und damit wahrscheinlich auch andere wertvolle Ketone und Terpene, von denen wir leider keine Daten haben) in der untersten Reihe rechts werden geschohnt.
Der Übersichtlichkeit halber sind die Säuren nicht dargestellt. Denn diese haben mit dem Vorlauf in jedem Fall nichts zu tun. Ebenfalls nicht dargestellt werden die höheren Alkohole. Diese würden in diesem Fall ohne Rektifikation im Schnitt zu 15% abgetrennt werden und mit Rektifikation so gut wie gar nicht. Hier ist ein kleiner Vorteil des Vorlaufabtrennens ohne Reflux.
Mit dem Rechner Begleitstoffe - Fraktionssimulator kann man das auch sehr gut veranschaulichen:
Dieser rechnet eine Destillation vollständig durch und zeigt dann Werte der Begleitstoffe an. Dabei kann man mit einem Schieberegler die Länge der angezeigten Fraktion einstellen und somit sehr schnell Vergleiche bezüglich der Fraktionslänge durchführen. Hier eine Tabelle über Vorlaufabtrennung mit verschieden hoher Rektifikation und verschieden hoher Alkoholstärke im Kessel:
Vier verscheidene Anfangsalkoholstärken und drei verschiedene Rektifikationsstufen. Abgetrennt wurde, sobald die Summe des Acetaldehyds und des Ethylacetats (also der beiden wichtigsten Vorlaufstoffen) 180% ergeben. Angezeigt werden, wie viel % des Alkohols man im Vorlauf verliert ("%Eth") und wieviel % der drei wetvollen Ester Ethylcaproat, Ethylcaprylat und Ethylcaprinat ("%C") im Schnitt (also die Summe der drei Werte durch drei geteilt), welche möglichst nicht im Vorlauf landen sollen.
Man sieht, daß man mit möglichst hoher Rektifikation die besten Ergebnisse bekommt (am wenigsten %Eth und am wenigsten %C im Vorlauf ist). Das ist keine Überraschung. Überraschend ist aber, daß hohe vol% im Kessel zur Folge haben, daß man mehr Alkohol verliert, um die geforderte Menge Acetaldehyd und Ethylacetat abzutrennen. Wenn man sich die Werte im Fraktionssimulator genauer anschaut, merkt man, daß das Problem das Ethylacetat ist. Dieses lässt sich generell sehr schlecht bei hohen Alkoholprozent im Kessel abtrennen. Also das Acetaldehyd lässt sich zwar sehr gut abtrennen bei hohen vol%, das Ethylacetat aber nicht. Allgemein lassen sich die Ester schlecht bei hohen vol% im Kessel abtrennen. Deswegen verliert man auch von den guten Estern mehr, wenn die vol% im Kessel niedrig sind. Bei hoher Rektifikation macht das aber nicht mehr viel aus.
Mit dem Begleitstoffesimulator 2 kann man sich sehr gut das Endergebnis anschauen, je nachdem, ob man beim Feinbrand den Vorlauf rektifiziert oder nicht:
Feinbrand mit 10lt 30vol%.
Da der Vorlauf rektifiziert natürlich viel konzentrierter ist, muss bei der Destillation mit Rektifikation weniger davon abgetrennt werden, damit die Ergebnisse am Ende vergleichbar sind:
A)
Vorlauf 0.2lt mit 1.5 theor. Böden. Dann Mittellauf bis aktuell 63vol% mit 1.2 theor. Böden. Der Mittellauf schaut dann so aus: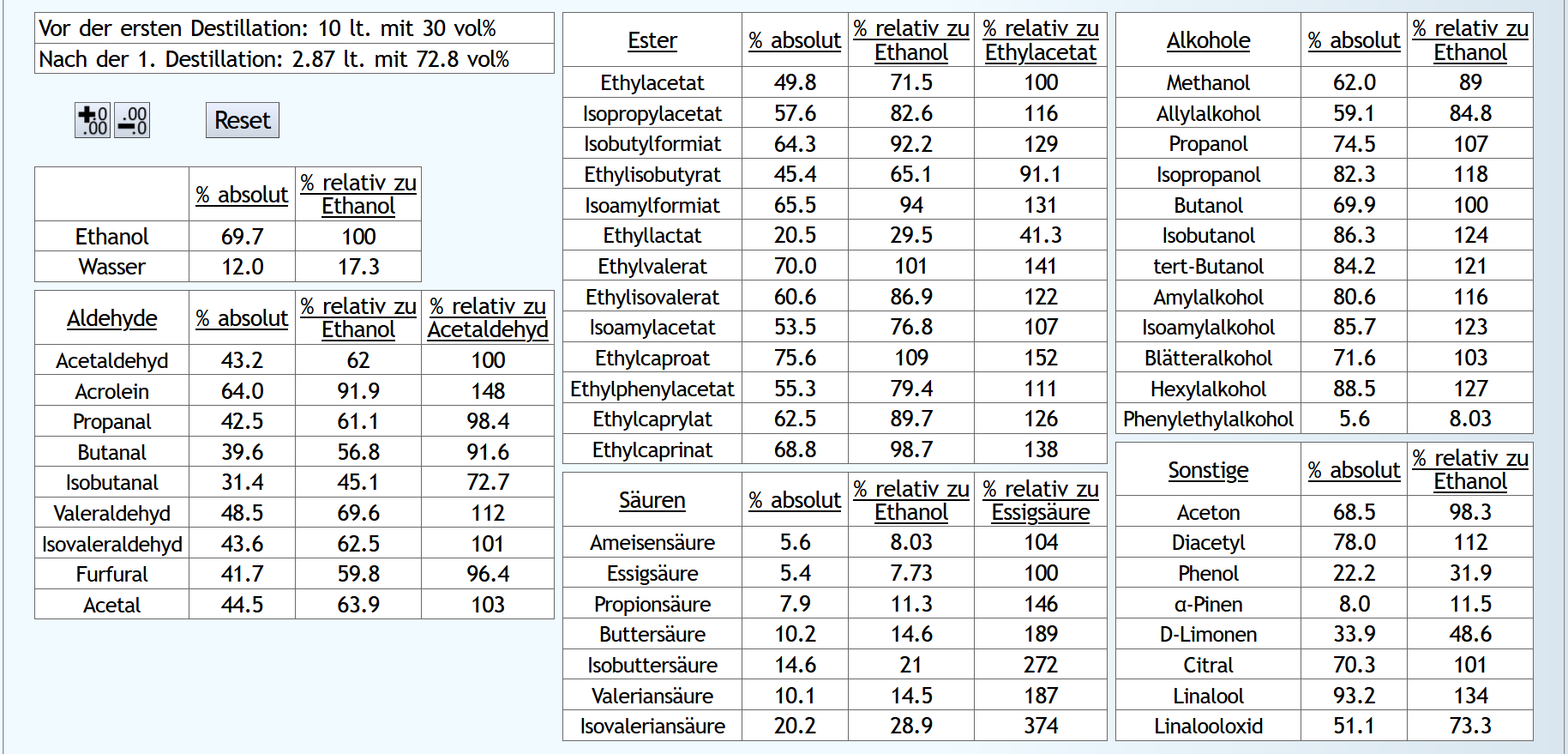 B)
B)
Vorlauf 0.02lt mit 6 theor. Böden. Dann Mittellauf bis aktuell 63vol% mit 1.2 theor. Böden. Der Mittellauf schaut dann so aus: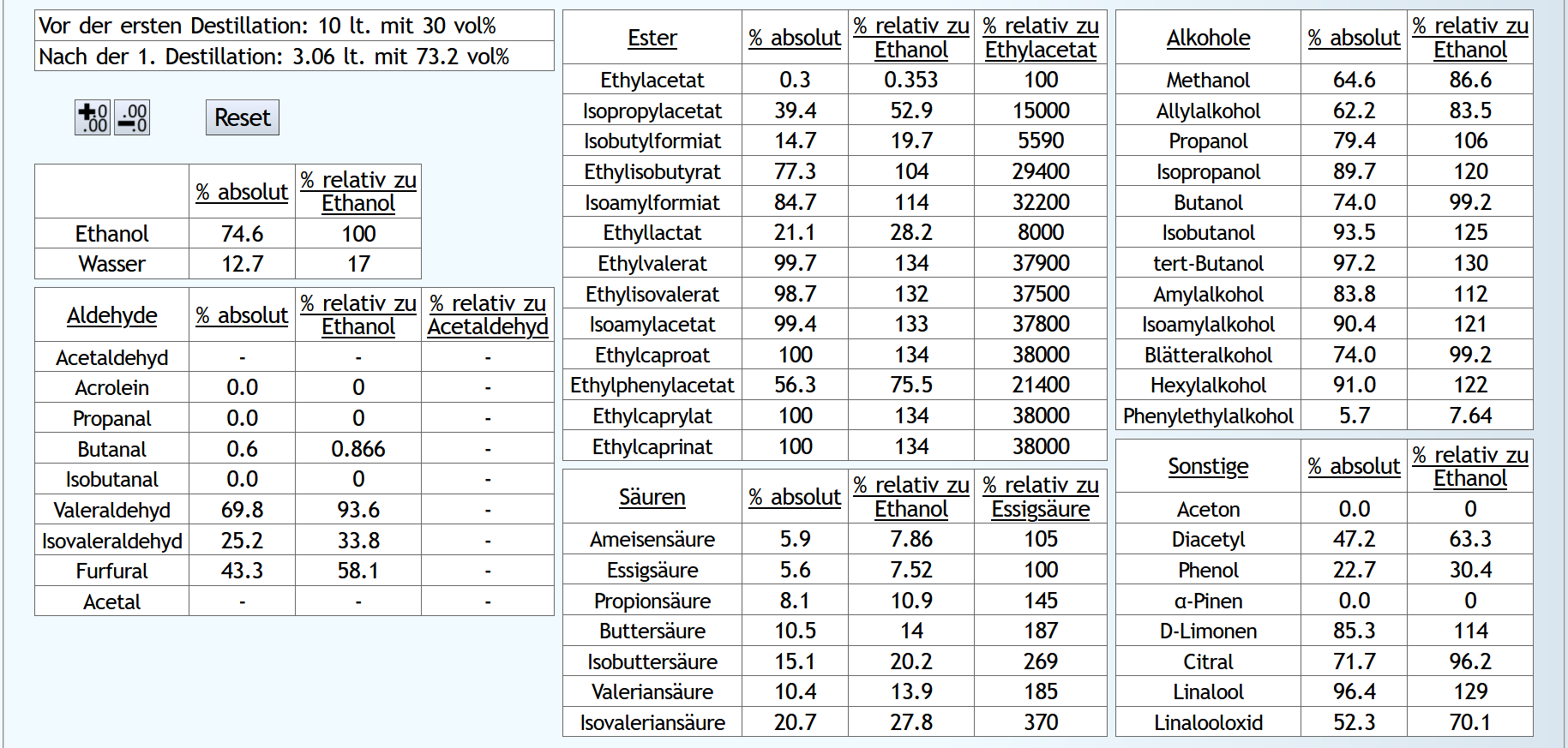 Mit Rektifikation sind die kleineren, schlechten Aldehyde fast vollkommen weg.
Von den wertvollen sind aber noch ähnliche Mengen da verglichen mit der Destillation ohne Rektifikation des Vorlaufs.
Mit Rektifikation ist Ethylacetat fast vollkommen weg, ohne nur die Hälfte.
Weiter unten die großen, guten Ester sind mit Rektifikation zum Teil komplett im Mittellauf gelandet, ohne Rektifikation nur im Schnitt so zu 60%.
Kaum Unterschiede bei den Säuren und Alkoholen.
Mit Rektifikation bekommt man Aceton weg.
Diacetyl ist halbiert.
Leider wird a-Pinene noch besser abgetrennt.
Aber die anderen Terpene werden zum Teil deutlich mehr in den Mittellauf gebracht.
Und natürlich hat man eine höhere Ethanolausbeute, wenn man den Vorlauf konzentrierter abtrennt.
Mit Rektifikation sind die kleineren, schlechten Aldehyde fast vollkommen weg.
Von den wertvollen sind aber noch ähnliche Mengen da verglichen mit der Destillation ohne Rektifikation des Vorlaufs.
Mit Rektifikation ist Ethylacetat fast vollkommen weg, ohne nur die Hälfte.
Weiter unten die großen, guten Ester sind mit Rektifikation zum Teil komplett im Mittellauf gelandet, ohne Rektifikation nur im Schnitt so zu 60%.
Kaum Unterschiede bei den Säuren und Alkoholen.
Mit Rektifikation bekommt man Aceton weg.
Diacetyl ist halbiert.
Leider wird a-Pinene noch besser abgetrennt.
Aber die anderen Terpene werden zum Teil deutlich mehr in den Mittellauf gebracht.
Und natürlich hat man eine höhere Ethanolausbeute, wenn man den Vorlauf konzentrierter abtrennt.
Also fast nur gutes bringt die Rektifikation Vorlaufs. Vor allem bei den Estern.
Das bedeutet auch, daß mit der Rektifikation des Vorlaufs Probleme gelöst werden können, welche bei Brennweisen, die bei anderen Aspekten, wie zum Beispiel der Menge an Säuren, Vorteile bieten, aber bei den Estern und Aldehyden nicht optimal sind. Also Potstill-Mehrfachdestillation. Also wenn man die Möglichkeit hat, den Vorlauf zu rektifizieren, kann man sich bei der Wahl der Brennweise auf andere Dinge konzentrieren.
Fazit: Insgesamt ist ganz klar ersichtlich, daß ein Vorlaufabtrennen mit Rektifikation nicht nur effektiver sondern auch qualitativ besser ist.
40vol%, 1 theor. Boden, Zoom x10:
Nun mit 6 theor. Böden:
Der Übersichtlichkeit halber sind die Säuren nicht dargestellt. Denn diese haben mit dem Vorlauf in jedem Fall nichts zu tun. Ebenfalls nicht dargestellt werden die höheren Alkohole. Diese würden in diesem Fall ohne Rektifikation im Schnitt zu 15% abgetrennt werden und mit Rektifikation so gut wie gar nicht. Hier ist ein kleiner Vorteil des Vorlaufabtrennens ohne Reflux.
Mit dem Rechner Begleitstoffe - Fraktionssimulator kann man das auch sehr gut veranschaulichen:
Dieser rechnet eine Destillation vollständig durch und zeigt dann Werte der Begleitstoffe an. Dabei kann man mit einem Schieberegler die Länge der angezeigten Fraktion einstellen und somit sehr schnell Vergleiche bezüglich der Fraktionslänge durchführen. Hier eine Tabelle über Vorlaufabtrennung mit verschieden hoher Rektifikation und verschieden hoher Alkoholstärke im Kessel:
| Kessel | 1 theor. Boden | 2 theor. Böden | 4 theor. Böden |
| 5vol% | 36.1%Eth 98.5%C | 7.1%Eth 97.0%C | 0.5%Eth 1.6%C |
| 15vol% | 32.7%Eth 98.0%C | 7.8%Eth 61.5%C | 0.7%Eth 0.4%C |
| 35vol% | 31.3%Eth 93.9%C | 12.2%Eth 18.7%C | 1.3%Eth 0.1%C |
| 60vol% | 37.5%Eth 34.2%C | 13.8%Eth 4.5%C | 2.2%Eth 0.0%C |
Vier verscheidene Anfangsalkoholstärken und drei verschiedene Rektifikationsstufen. Abgetrennt wurde, sobald die Summe des Acetaldehyds und des Ethylacetats (also der beiden wichtigsten Vorlaufstoffen) 180% ergeben. Angezeigt werden, wie viel % des Alkohols man im Vorlauf verliert ("%Eth") und wieviel % der drei wetvollen Ester Ethylcaproat, Ethylcaprylat und Ethylcaprinat ("%C") im Schnitt (also die Summe der drei Werte durch drei geteilt), welche möglichst nicht im Vorlauf landen sollen.
Man sieht, daß man mit möglichst hoher Rektifikation die besten Ergebnisse bekommt (am wenigsten %Eth und am wenigsten %C im Vorlauf ist). Das ist keine Überraschung. Überraschend ist aber, daß hohe vol% im Kessel zur Folge haben, daß man mehr Alkohol verliert, um die geforderte Menge Acetaldehyd und Ethylacetat abzutrennen. Wenn man sich die Werte im Fraktionssimulator genauer anschaut, merkt man, daß das Problem das Ethylacetat ist. Dieses lässt sich generell sehr schlecht bei hohen Alkoholprozent im Kessel abtrennen. Also das Acetaldehyd lässt sich zwar sehr gut abtrennen bei hohen vol%, das Ethylacetat aber nicht. Allgemein lassen sich die Ester schlecht bei hohen vol% im Kessel abtrennen. Deswegen verliert man auch von den guten Estern mehr, wenn die vol% im Kessel niedrig sind. Bei hoher Rektifikation macht das aber nicht mehr viel aus.
Mit dem Begleitstoffesimulator 2 kann man sich sehr gut das Endergebnis anschauen, je nachdem, ob man beim Feinbrand den Vorlauf rektifiziert oder nicht:
Feinbrand mit 10lt 30vol%.
Da der Vorlauf rektifiziert natürlich viel konzentrierter ist, muss bei der Destillation mit Rektifikation weniger davon abgetrennt werden, damit die Ergebnisse am Ende vergleichbar sind:
A)
Vorlauf 0.2lt mit 1.5 theor. Böden. Dann Mittellauf bis aktuell 63vol% mit 1.2 theor. Böden. Der Mittellauf schaut dann so aus:
Vorlauf 0.02lt mit 6 theor. Böden. Dann Mittellauf bis aktuell 63vol% mit 1.2 theor. Böden. Der Mittellauf schaut dann so aus:
Also fast nur gutes bringt die Rektifikation Vorlaufs. Vor allem bei den Estern.
Das bedeutet auch, daß mit der Rektifikation des Vorlaufs Probleme gelöst werden können, welche bei Brennweisen, die bei anderen Aspekten, wie zum Beispiel der Menge an Säuren, Vorteile bieten, aber bei den Estern und Aldehyden nicht optimal sind. Also Potstill-Mehrfachdestillation. Also wenn man die Möglichkeit hat, den Vorlauf zu rektifizieren, kann man sich bei der Wahl der Brennweise auf andere Dinge konzentrieren.
Fazit: Insgesamt ist ganz klar ersichtlich, daß ein Vorlaufabtrennen mit Rektifikation nicht nur effektiver sondern auch qualitativ besser ist.
Wie weit soll man Raubrände runterbrennen?
Die Frage ist, ob es geschmacklich Sinn macht, Raubrände über den Punkt, an dem kein Alkohol mehr kommt, zu verlängern.
Oder, im Gegenteil, wegen der Qualität schon aufzuhören, obwohl es sich bezüglich der Alkoholausbeute noch lohnen würde, weiter zu destillieren.
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 2 berechnet, welcher mehrere Destillationen hintereinander berechnen kann und die Ergebnisse als Tabellen ausgibt.
Schuld am schlechten Geruch und Geschmack von Nachlauf sind vor allem Carbonsäuren. Sie sind etwas, was man eigentlich möglichst wenig im Schnapsglas haben möchte. Andererseits sind sie Vorläufer von Estern. Und je größer die Säuremoleküle, desto besser die Ester. Auch die Säuren sind in den Rechnern nach der Molekülgröße angeordnet. Also gilt, daß die Säuren, welche im Simulator weiter unten stehen, die wertvolleren sind. Das stimmt aber nur bezüglich ihres Potentials bei der Veresterung. Ganz oben die Ameisensäure kommt recht wenig vor, die zweitoberste, die Essigsäure, ist dagegen bei weitem die häufigste. Diese beiden oberen werden hier nun als die schlechten angesehen. Die weiter unten als die guten, obwohl sie unverestert ganz klar dem Geschmack schaden. Wenn Säuren irgendwann zwischen dem Rau- und Feinbrand oder während des Feinbrands verestern, landen diese Ester größtenteils im Vorlauf und im Mittellauf. Und die Säuren, welche nicht verestern, werden beim Feinbrand dann möglichst vollständig als Nachlauf abgetrennt. Das ist jedenfalls der Plan. Nur wenn man die Säuren beim Feinbrand gut abtrennen kann, hat es Sinn, bei den Raubränden mehr davon zu sammeln.
10lt Maische mit 8vol%:
A)
Raubrand bis 2.5l, also ein eher kurzer Raubrand, mit 1.1 theor. Böden, also recht schnell mit isolierter Destille. Dann beim Feinbrand 0.03lt mit 1.5 theor. Böden Vorlauf, Mittellauf mit 1.3 theor. Böden bis 65vol% aktuellem Alkoholgehalt: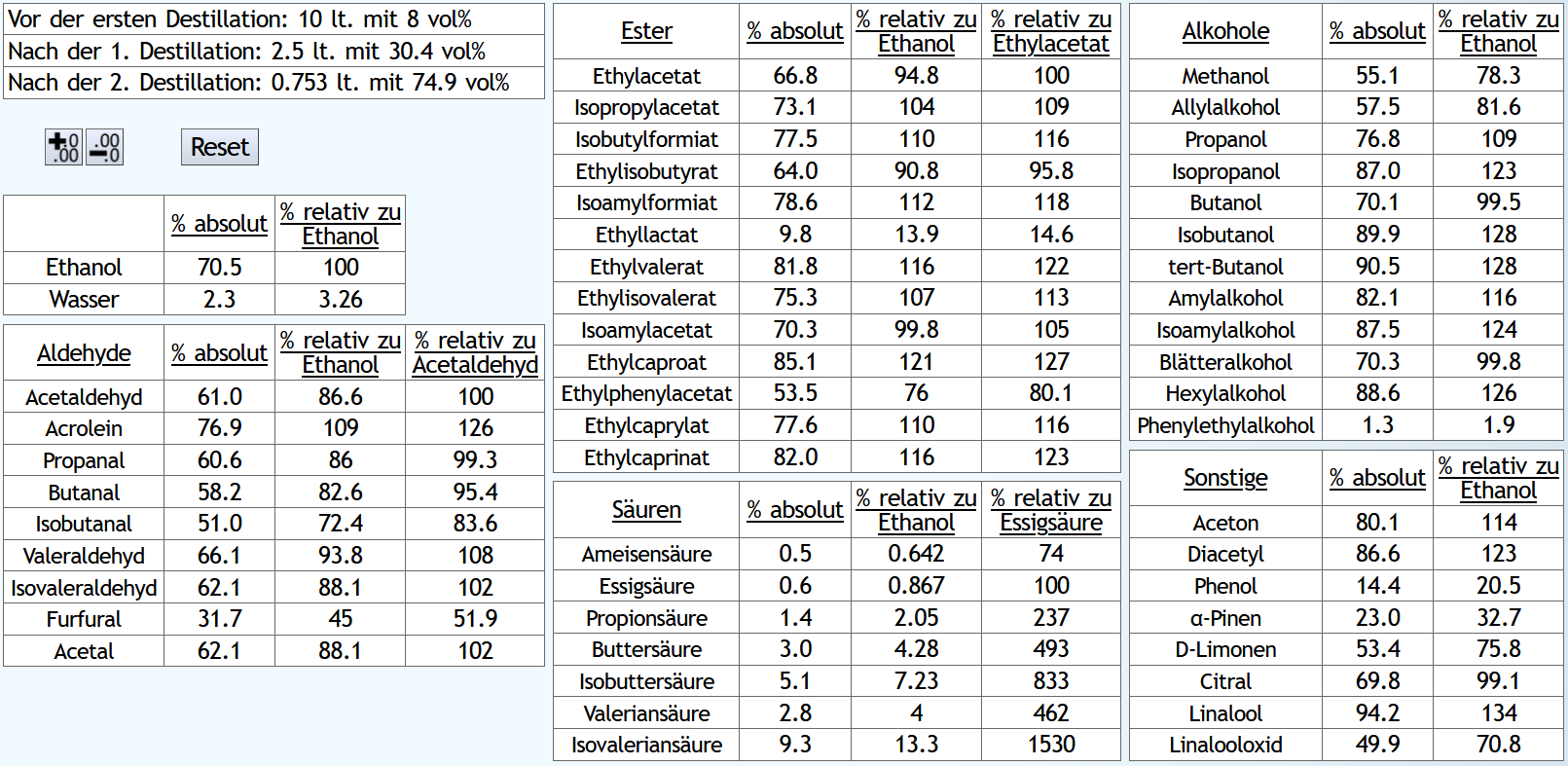 B)
B)
Raubrand bis 3.5l, also ein langer Raubrand, ebenfalls mit 1.1 theor. Böden. Dann beim Feinbrand der gleiche Vorlauf, Mittellauf bis 60vol% aktuellem Alkoholgehalt. 60 anstelle 65vol% Mittellaufende, da der Alkoholgehalt des Raubrands niedriger ist: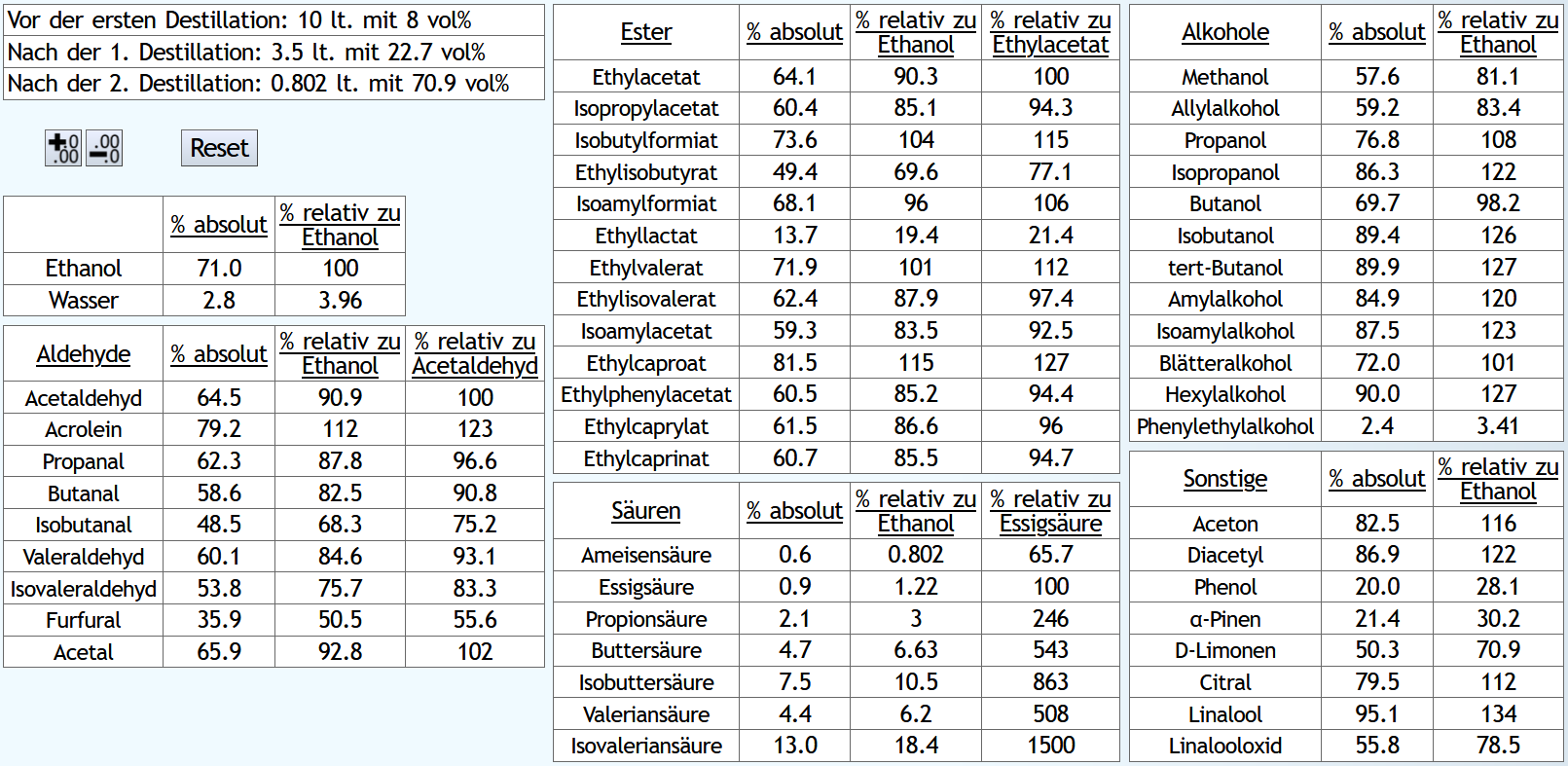 Der kurze Raubrand bewirkt am Ende weniger schlechte und mehr gute Aldehyde.
Und mehr von den guten Estern.
Etwas weniger höhere Alkohole und deutlich weniger Säuren.
Er ist also sauberer trotz höherer Alkoholausbeute.
Es schaut also so aus, als bekommt man beim Doppelbrand mit langem Raubrennen mehr Geschmack, aber tendenziell etwas mehr schlechte Geschmäcker als gute.
Der kurze Raubrand bewirkt am Ende weniger schlechte und mehr gute Aldehyde.
Und mehr von den guten Estern.
Etwas weniger höhere Alkohole und deutlich weniger Säuren.
Er ist also sauberer trotz höherer Alkoholausbeute.
Es schaut also so aus, als bekommt man beim Doppelbrand mit langem Raubrennen mehr Geschmack, aber tendenziell etwas mehr schlechte Geschmäcker als gute.
Der lange Raubrand bewirkt aber, daß man am Ende anteilig ein bisschen mehr von den großen, interessanten Säuren bekommt. Hier ist also ein besseres Potential zur Veresterung, allerdings auch die Gefahr, daß der Brand am Ende zu harsch wird, selbst wenn man den Mittellauf relativ früh beendet.
Interessant ist vielleicht noch Phenol: Dies ist bei dem langen Raubrand deutlich mehr vorhanden, 28.1 anstelle 20.5% im Vergleich zu Ethanol. Bei einer Maische mit getorftem Malz bewirkt ein langer Raubrand also einen höheren Rauchgehalt im Whisky.
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 2 berechnet, welcher mehrere Destillationen hintereinander berechnen kann und die Ergebnisse als Tabellen ausgibt.
Schuld am schlechten Geruch und Geschmack von Nachlauf sind vor allem Carbonsäuren. Sie sind etwas, was man eigentlich möglichst wenig im Schnapsglas haben möchte. Andererseits sind sie Vorläufer von Estern. Und je größer die Säuremoleküle, desto besser die Ester. Auch die Säuren sind in den Rechnern nach der Molekülgröße angeordnet. Also gilt, daß die Säuren, welche im Simulator weiter unten stehen, die wertvolleren sind. Das stimmt aber nur bezüglich ihres Potentials bei der Veresterung. Ganz oben die Ameisensäure kommt recht wenig vor, die zweitoberste, die Essigsäure, ist dagegen bei weitem die häufigste. Diese beiden oberen werden hier nun als die schlechten angesehen. Die weiter unten als die guten, obwohl sie unverestert ganz klar dem Geschmack schaden. Wenn Säuren irgendwann zwischen dem Rau- und Feinbrand oder während des Feinbrands verestern, landen diese Ester größtenteils im Vorlauf und im Mittellauf. Und die Säuren, welche nicht verestern, werden beim Feinbrand dann möglichst vollständig als Nachlauf abgetrennt. Das ist jedenfalls der Plan. Nur wenn man die Säuren beim Feinbrand gut abtrennen kann, hat es Sinn, bei den Raubränden mehr davon zu sammeln.
10lt Maische mit 8vol%:
A)
Raubrand bis 2.5l, also ein eher kurzer Raubrand, mit 1.1 theor. Böden, also recht schnell mit isolierter Destille. Dann beim Feinbrand 0.03lt mit 1.5 theor. Böden Vorlauf, Mittellauf mit 1.3 theor. Böden bis 65vol% aktuellem Alkoholgehalt:
Raubrand bis 3.5l, also ein langer Raubrand, ebenfalls mit 1.1 theor. Böden. Dann beim Feinbrand der gleiche Vorlauf, Mittellauf bis 60vol% aktuellem Alkoholgehalt. 60 anstelle 65vol% Mittellaufende, da der Alkoholgehalt des Raubrands niedriger ist:
Der lange Raubrand bewirkt aber, daß man am Ende anteilig ein bisschen mehr von den großen, interessanten Säuren bekommt. Hier ist also ein besseres Potential zur Veresterung, allerdings auch die Gefahr, daß der Brand am Ende zu harsch wird, selbst wenn man den Mittellauf relativ früh beendet.
Interessant ist vielleicht noch Phenol: Dies ist bei dem langen Raubrand deutlich mehr vorhanden, 28.1 anstelle 20.5% im Vergleich zu Ethanol. Bei einer Maische mit getorftem Malz bewirkt ein langer Raubrand also einen höheren Rauchgehalt im Whisky.
Wie weit soll man beim Feinbrand Nachlauf sammeln?
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 2 berechnet.
30vol% im Kessel, Feinbrand mit 1.3 theor. Böden, Mittellaufende bei 65vol% im Dampf. Dann Nachlaufsammeln mit 1 theor. Boden nur bis 20vol% im Dampf. Der Säuren im Nachlauf schauen dann so aus: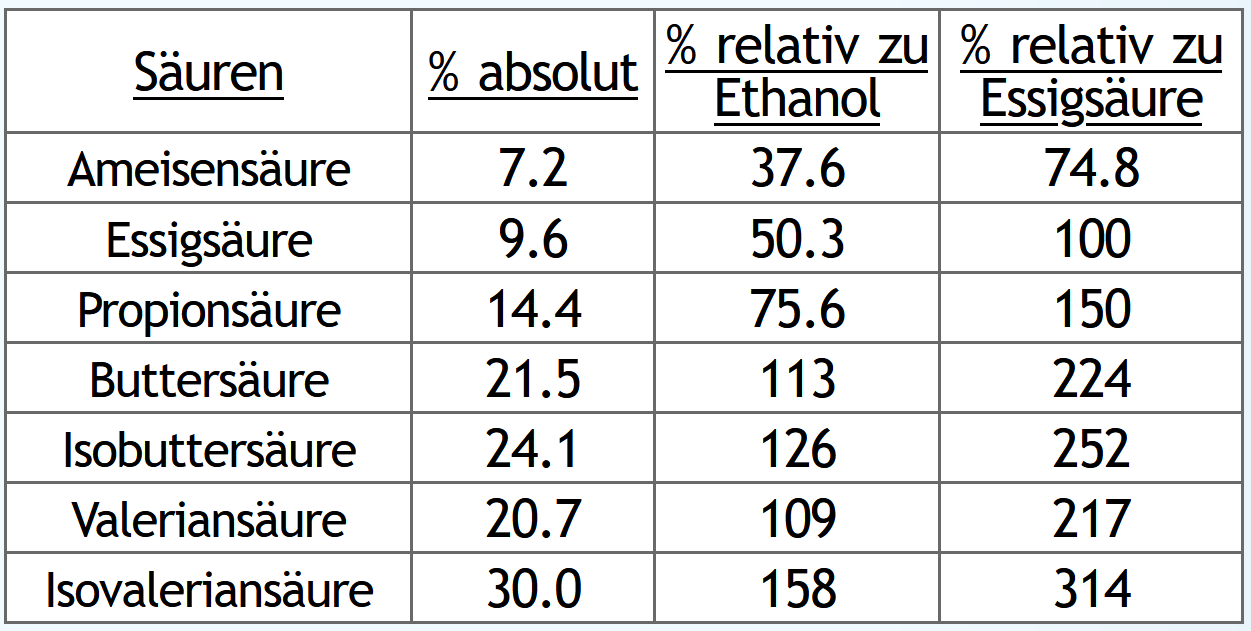 Nun das gleiche, aber Nachlauf gesammelt bis 5vol% im Dampf:
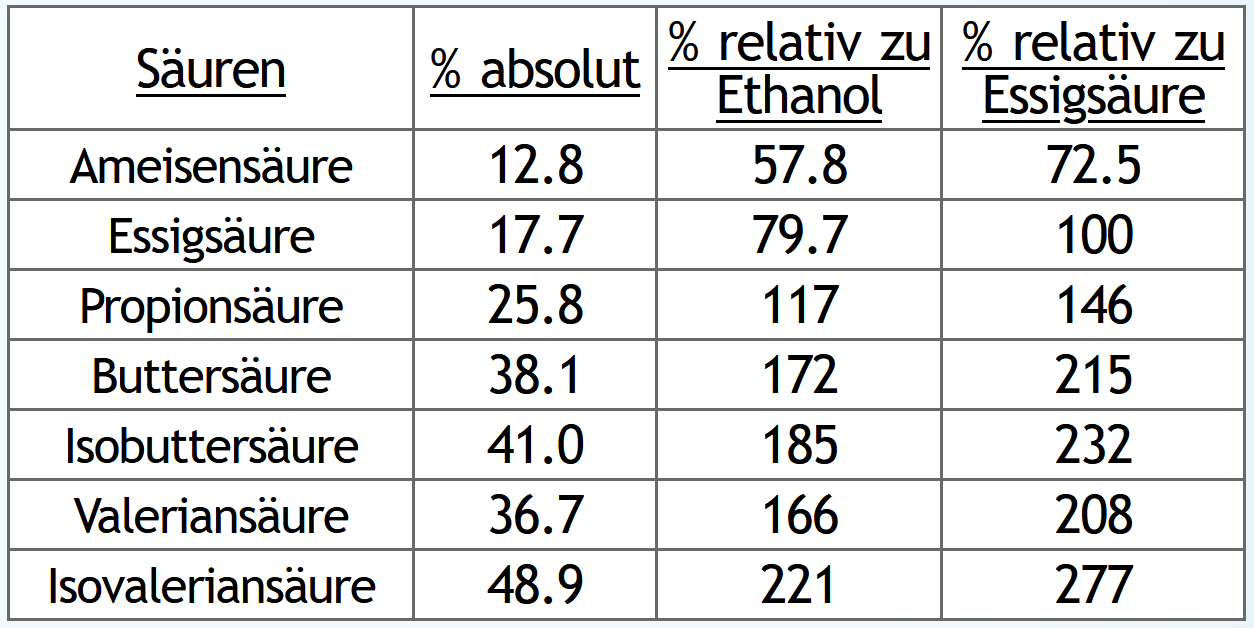
Nun das gleiche, aber Nachlauf gesammelt bis 5vol% im Dampf:
 Und nun das gleiche, aber Nachlauf gesammelt bis 0.1vol% im Dampf:
Und nun das gleiche, aber Nachlauf gesammelt bis 0.1vol% im Dampf:
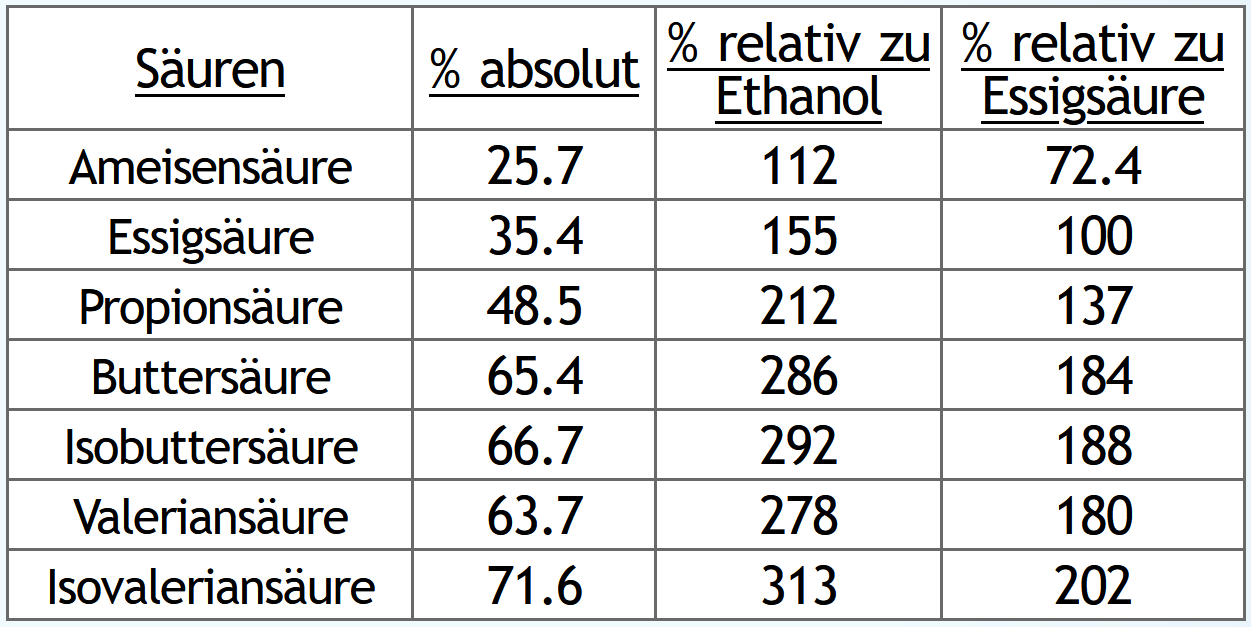 Wie erwartet steigen natürlich die absouten Mengen an Säuren, wenn man länger Nachlauf sammelt.
Und auch wie erwartet steigen die Säuren relativ zur Ethanolausbeute.
Spannender ist jeweils die dritte Tabellenspalte "% relativ zur Essigsäure":
Hier sieht man, wie das Verhältnis der Säuren untereinander sich verändert.
Je länger man Nachlauf sammelt, desto anteilig mehr bekommt man Säuren mit kleiner Molekülmasse.
Also zum Beispiel immer weniger Valerian- und Isovaleriansäure und dafür immer mehr Essigsäure.
Die größeren Säuren haben das Potential, sehr interessantere Ester zu bilden.
Die kleine Essigsäure dagegen vor allem nur Ethylacetat, also einen klebstoffähnlichen Geruch.
Wie erwartet steigen natürlich die absouten Mengen an Säuren, wenn man länger Nachlauf sammelt.
Und auch wie erwartet steigen die Säuren relativ zur Ethanolausbeute.
Spannender ist jeweils die dritte Tabellenspalte "% relativ zur Essigsäure":
Hier sieht man, wie das Verhältnis der Säuren untereinander sich verändert.
Je länger man Nachlauf sammelt, desto anteilig mehr bekommt man Säuren mit kleiner Molekülmasse.
Also zum Beispiel immer weniger Valerian- und Isovaleriansäure und dafür immer mehr Essigsäure.
Die größeren Säuren haben das Potential, sehr interessantere Ester zu bilden.
Die kleine Essigsäure dagegen vor allem nur Ethylacetat, also einen klebstoffähnlichen Geruch.
Fazit: Wenn man den Nachlauf sammelt, um ihn irgendwann als Aromabooster einzusetzen, bewirkt ein langes Nachlaufsammeln, daß man zwar sehr viele Säuren sammelt, also schlussendlich das Aroma dann auch sehr stark gefördert wird, aber eher dann mit uninteressanteren Aromen. Was aber natürlich auch eine Geschmacksfrage ist.
Beim folgenden Feinbrand mit dem Nachlauf im Kessel kann man aber mit einer Rektifikation des Vorlaufs sehr gut die schlechten Ester abtrennen. Und mit einer recht hohen Anfangsalkoholstärke im Kessel und einem relativ frühen Nachlaufschnitt auch die überschüssigen Säuren. So kann man die Vorteile der großen Anzahl an Säuren durch das lange Nachlaufsammeln mitnehmen und die entstehenden Nachteile reduzieren.
Geht es beim Nachlaufsammeln nur darum, den Alkohol irgendwann neutral zu brennen, dann entscheiden der höhere Aufwand, ihn zu sammeln und von der Mehrmenge an Säuren zu reinigen, darüber, wie lange man brennen sollte.
30vol% im Kessel, Feinbrand mit 1.3 theor. Böden, Mittellaufende bei 65vol% im Dampf. Dann Nachlaufsammeln mit 1 theor. Boden nur bis 20vol% im Dampf. Der Säuren im Nachlauf schauen dann so aus:
Fazit: Wenn man den Nachlauf sammelt, um ihn irgendwann als Aromabooster einzusetzen, bewirkt ein langes Nachlaufsammeln, daß man zwar sehr viele Säuren sammelt, also schlussendlich das Aroma dann auch sehr stark gefördert wird, aber eher dann mit uninteressanteren Aromen. Was aber natürlich auch eine Geschmacksfrage ist.
Beim folgenden Feinbrand mit dem Nachlauf im Kessel kann man aber mit einer Rektifikation des Vorlaufs sehr gut die schlechten Ester abtrennen. Und mit einer recht hohen Anfangsalkoholstärke im Kessel und einem relativ frühen Nachlaufschnitt auch die überschüssigen Säuren. So kann man die Vorteile der großen Anzahl an Säuren durch das lange Nachlaufsammeln mitnehmen und die entstehenden Nachteile reduzieren.
Geht es beim Nachlaufsammeln nur darum, den Alkohol irgendwann neutral zu brennen, dann entscheiden der höhere Aufwand, ihn zu sammeln und von der Mehrmenge an Säuren zu reinigen, darüber, wie lange man brennen sollte.
Gibt es eine wertvolle Fraktion nach dem Nachlauf?
Immer mal wieder liest man, daß nach dem Nachlauf noch eine Fraktion gesammelt wird.
Diese kann dann als Verdünnungswasser verwendet werden.
Dafür muss sie natürlich besser sein als der davor abdestillierte Nachlauf.
Diese Fraktion wird oft "sweet water" oder z.B. in Arroyos Schriften über die Rumherstellung "fifth fraction" genannt (foreshots, heads, hearts und tails sind die Fraktionen 1-4).
Diese Fraktion soll besser sein als die davor, vor allem süßer, oder vielleicht ist weniger sauer gemeint.
Das, was am Nachlauf so schlecht schmeckt, sind vor allem Säuren wie Essigsäure oder Buttersäure. Die Frage ist nun, ob diese Säuren irgendwann abdestilliert sind, sodaß danach ein säurefreises Destillat kommt. Wenn nicht, dann ist eine späte Fraktion, welche man wirklich direkt ins Endprodukt mischen kann, undenkbar.
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 1 berechnet.
30vol%, 1 theor. Boden: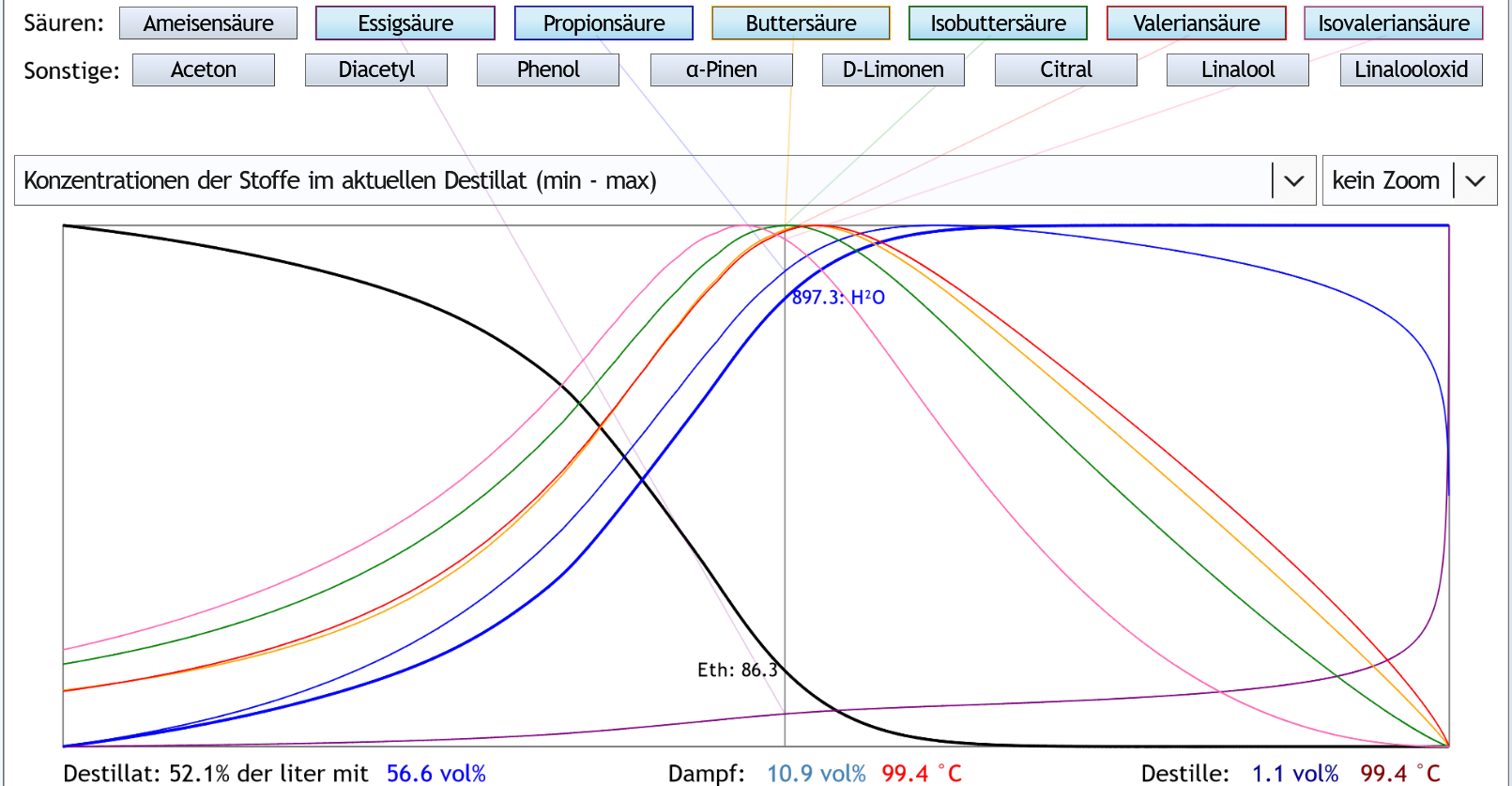 Der Peak der Säuren mt hoher Molekülmasse liegt in einem Bereich, wo noch Alkohol im Dampf ist.
So etwa bei 11vol% im Dampf.
Normalerweise ist das beim Nachlauf.
Und die Essigsäure (kleine Molekülmasse) steigt langsam aber stetig an.
Die y-Achse dieses Diagramms geht vom berechneten Minimalwert bis zum berechneten Maximalwert dieses Stoffes.
Jeder Stoff hat also eine andere y-Achse.
Essigsäure macht ja normalerweise bei weitem den Hauptteil der Säuren aus.
Deswegen bedeutet das langsame Ansteigen der Essigsäure eine größere Säuerung des Destillats als das Wegfallen der großen Säuren eine Verringerung der Säuerung bedeutet.
Daß das Destillat nach dem Peak weniger sauer wird, kann man also nicht so sagen.
Wohl kann man sagen, daß die großen Säuren in der Fraktion nach dem Nachlauf weniger vertreten sind als davor.
Also deren penetranter Geruch (viel schlimmer als der von Essigsäure) findet sich vor allem gegen Ende des Nachlaufs wieder, nicht im Destillat danach.
Der Peak der Säuren mt hoher Molekülmasse liegt in einem Bereich, wo noch Alkohol im Dampf ist.
So etwa bei 11vol% im Dampf.
Normalerweise ist das beim Nachlauf.
Und die Essigsäure (kleine Molekülmasse) steigt langsam aber stetig an.
Die y-Achse dieses Diagramms geht vom berechneten Minimalwert bis zum berechneten Maximalwert dieses Stoffes.
Jeder Stoff hat also eine andere y-Achse.
Essigsäure macht ja normalerweise bei weitem den Hauptteil der Säuren aus.
Deswegen bedeutet das langsame Ansteigen der Essigsäure eine größere Säuerung des Destillats als das Wegfallen der großen Säuren eine Verringerung der Säuerung bedeutet.
Daß das Destillat nach dem Peak weniger sauer wird, kann man also nicht so sagen.
Wohl kann man sagen, daß die großen Säuren in der Fraktion nach dem Nachlauf weniger vertreten sind als davor.
Also deren penetranter Geruch (viel schlimmer als der von Essigsäure) findet sich vor allem gegen Ende des Nachlaufs wieder, nicht im Destillat danach.
Andere Darstellung: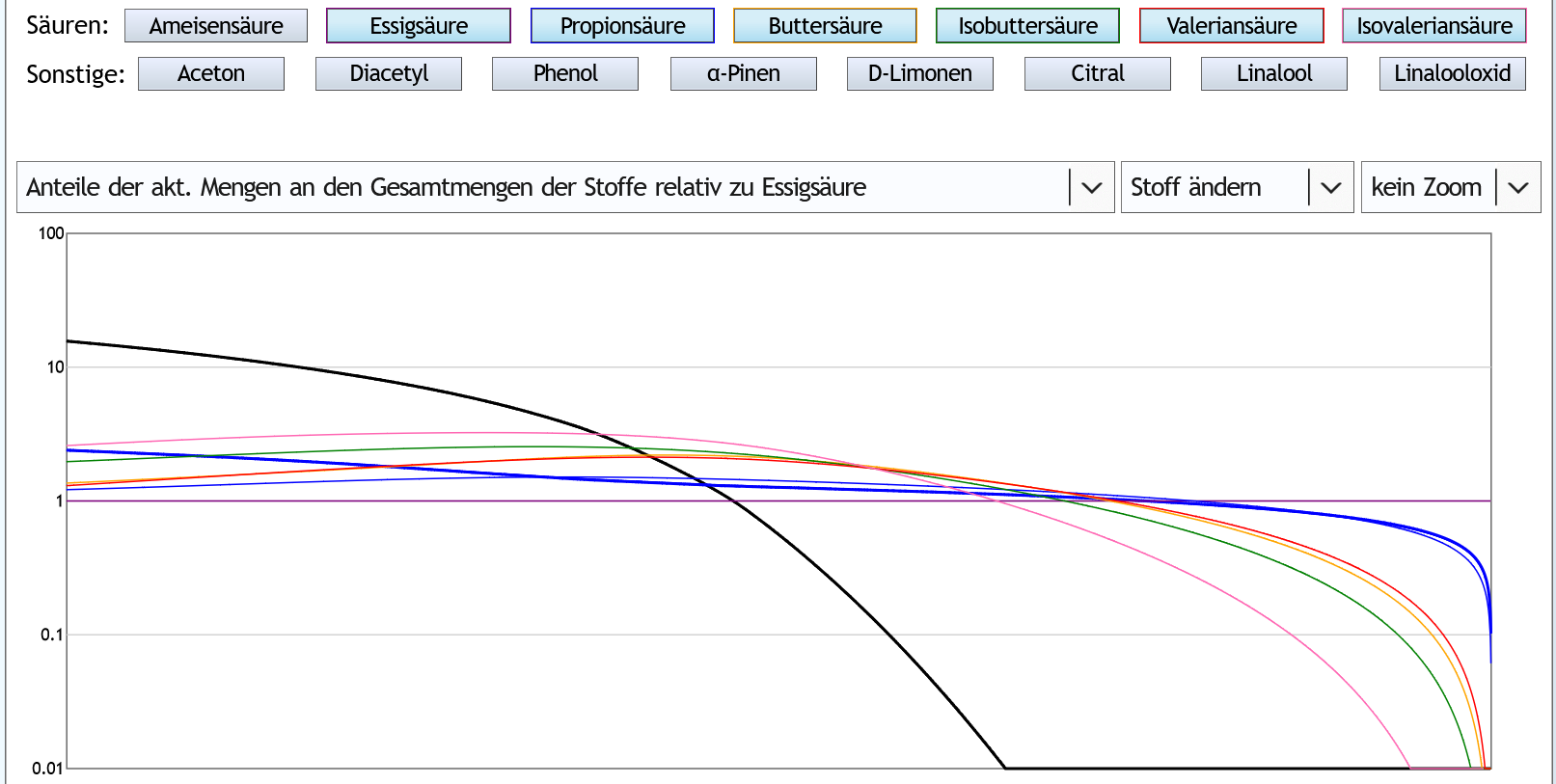 Hier sieht man das Verhältnis zwischen aktueller Essigsäure und den anderen Säuren.
Der Abstand wird zuerst langsam größer.
Bei etwa 50vol% im Dampf, also wahrscheinlich recht am Anfang des Nachlaufs, ist er am größten.
Dann nähern sich die Werte an.
Und etwa, wenn man 70% des Destilleninhalts destilliert hat, also in der Fraktion nach dem Nachlauf, kreuzen die Kurven die Linie von der Essigsäure.
Ab da sammelt man also anteilig weniger große Säuren als Essigsäure verglichen mit den Verhältnissen vor der Destillation.
Hier sieht man das Verhältnis zwischen aktueller Essigsäure und den anderen Säuren.
Der Abstand wird zuerst langsam größer.
Bei etwa 50vol% im Dampf, also wahrscheinlich recht am Anfang des Nachlaufs, ist er am größten.
Dann nähern sich die Werte an.
Und etwa, wenn man 70% des Destilleninhalts destilliert hat, also in der Fraktion nach dem Nachlauf, kreuzen die Kurven die Linie von der Essigsäure.
Ab da sammelt man also anteilig weniger große Säuren als Essigsäure verglichen mit den Verhältnissen vor der Destillation.
Nun mal schauen, ob etwas Rektifikation was daran ändert. 1.3 theor.Böden: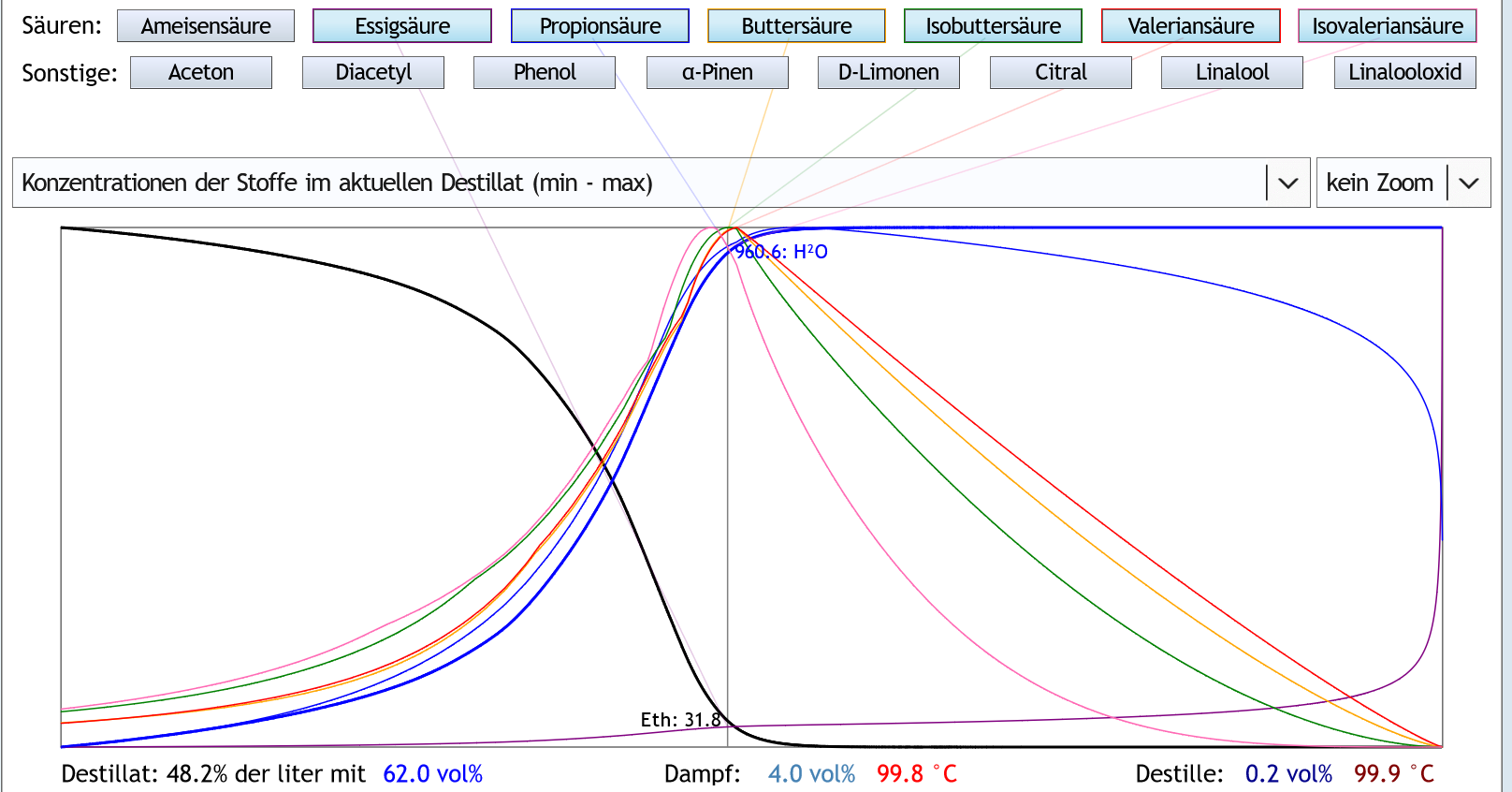
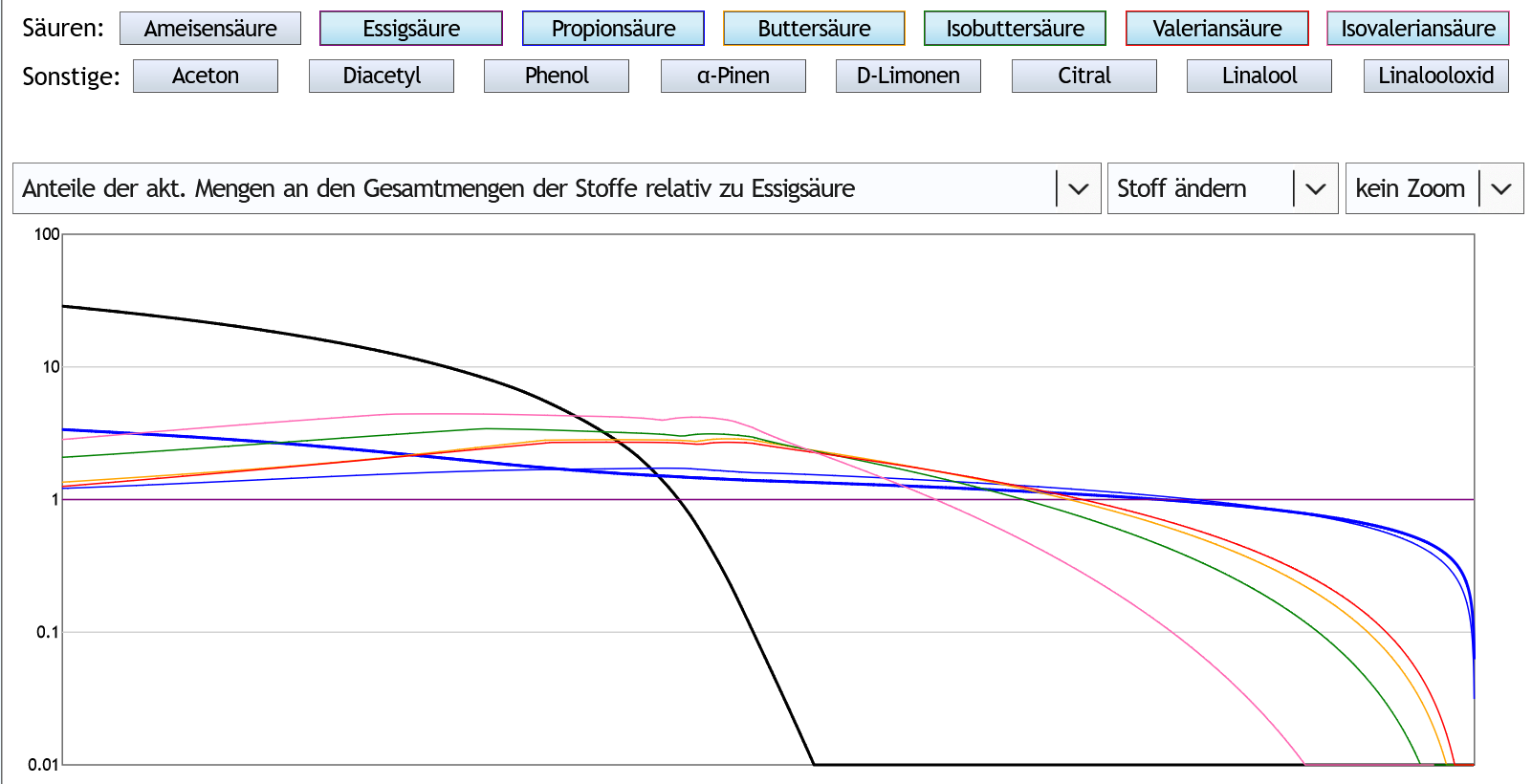 Der Peak der großen Säuren ist spitzer und später, bei etwa 4vol% im Dampf.
Das ändert aber im Prinzip nicht viel.
Nur der Punkt, ab dem die großen Säuren auftauchen, ist später, und der Punkt, ab dem sie wieder verschwinden, ist früher.
Im Bezug auf die Fragestellung bedeutet das, daß Rektifikation die mit großen Säuren belastete Fraktion komprimiert.
Also sie ist ungeniesbarer, aber kürzer.
Der Peak der großen Säuren ist spitzer und später, bei etwa 4vol% im Dampf.
Das ändert aber im Prinzip nicht viel.
Nur der Punkt, ab dem die großen Säuren auftauchen, ist später, und der Punkt, ab dem sie wieder verschwinden, ist früher.
Im Bezug auf die Fragestellung bedeutet das, daß Rektifikation die mit großen Säuren belastete Fraktion komprimiert.
Also sie ist ungeniesbarer, aber kürzer.
Zusammenfassend kann man vielleicht sagen, daß es eine Fraktion nach dem Nachlauf gibt, welche grundsätzlich anders ist als die davor, weil sie nämlich eine Entwicklung zeigt, welche vorher nicht da war: das Verschwinden der großen Säuren. Das bedeutet ein Verschwinden von sehr unangenehmen Gerüchen. Diese Fraktion ist sehr spät. Die Bezeichnung "sweet water" ist aber irreführend. Denn das stetige Ansteigen der Essigsäuremenge bedeutet ein stetiges Ansteigen des Säuregehalts, bis der Kessel leergebrannt ist. Vielleicht lässt uns der bessere Geruch der Fraktion nach dem Nachlauf denken, auch der Geschmack sei nun besser.
Allerdings: wegen der allgemeinen Regel, daß die Flüchtigkeit der Begleitstoffe in Wasser höher ist als in Alkohol, ist es unmöglich, daß ein Stoff, der beim Feinbrand ganz spät ins Destillat kommt, dann im Glas (also bei Trink-Alkoholstärke) einen deutlich wahrnehmbaren Geruch hat. Der Alkohol sorgt dafür, daß die Säuren im Schnaps bleiben, also nicht in die Nase kommen. Die Fraktion nach dem Nachlauf kann dem Schnaps also nur einen von der Nase nicht wahrgenommenen Geschmack oder vielleicht auch nur ein Mundgefühl hinzufügen.
Und Langzeiteffekte, nämlich daß bei der Lagerung Säuren verestern, ist eher ein Argument gegen das Verwenden der Fraktion nach dem Nachlauf. Denn die in dieser Fraktion vorherrschende Essigsäure bildet nicht so interessante Ester wie die mit dem Nachlauf verworfenen größeren Säuren.
Das, was am Nachlauf so schlecht schmeckt, sind vor allem Säuren wie Essigsäure oder Buttersäure. Die Frage ist nun, ob diese Säuren irgendwann abdestilliert sind, sodaß danach ein säurefreises Destillat kommt. Wenn nicht, dann ist eine späte Fraktion, welche man wirklich direkt ins Endprodukt mischen kann, undenkbar.
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 1 berechnet.
30vol%, 1 theor. Boden:
Andere Darstellung:
Nun mal schauen, ob etwas Rektifikation was daran ändert. 1.3 theor.Böden:
Zusammenfassend kann man vielleicht sagen, daß es eine Fraktion nach dem Nachlauf gibt, welche grundsätzlich anders ist als die davor, weil sie nämlich eine Entwicklung zeigt, welche vorher nicht da war: das Verschwinden der großen Säuren. Das bedeutet ein Verschwinden von sehr unangenehmen Gerüchen. Diese Fraktion ist sehr spät. Die Bezeichnung "sweet water" ist aber irreführend. Denn das stetige Ansteigen der Essigsäuremenge bedeutet ein stetiges Ansteigen des Säuregehalts, bis der Kessel leergebrannt ist. Vielleicht lässt uns der bessere Geruch der Fraktion nach dem Nachlauf denken, auch der Geschmack sei nun besser.
Allerdings: wegen der allgemeinen Regel, daß die Flüchtigkeit der Begleitstoffe in Wasser höher ist als in Alkohol, ist es unmöglich, daß ein Stoff, der beim Feinbrand ganz spät ins Destillat kommt, dann im Glas (also bei Trink-Alkoholstärke) einen deutlich wahrnehmbaren Geruch hat. Der Alkohol sorgt dafür, daß die Säuren im Schnaps bleiben, also nicht in die Nase kommen. Die Fraktion nach dem Nachlauf kann dem Schnaps also nur einen von der Nase nicht wahrgenommenen Geschmack oder vielleicht auch nur ein Mundgefühl hinzufügen.
Und Langzeiteffekte, nämlich daß bei der Lagerung Säuren verestern, ist eher ein Argument gegen das Verwenden der Fraktion nach dem Nachlauf. Denn die in dieser Fraktion vorherrschende Essigsäure bildet nicht so interessante Ester wie die mit dem Nachlauf verworfenen größeren Säuren.
Neutral-Doppelbrand mit oder ohne Verdünnen zwischen den Bränden?
Das folgende Beispiel ist mit dem Begleitstoffesimulator 2 berechnet.
10lt mit 8vol%.
Um ein deutliches Ergebnis zu bekommen, wird nur wenig Vorlauf abgetrennt, nicht allzustark rektifiziert und recht spät der Mittellauf beendet.
A)
Raubrand: bis 2.5lt mit 1.1 theor. Böden.
Feinbrand: 0.03lt Vorlauf mit 6 theor. Böden. Dann Mittellauf bis 90% des Alkohols destilliert ist; ebenfalls mit 6 theor. Böden.
Das Rechenergebnis für den Mittellauf: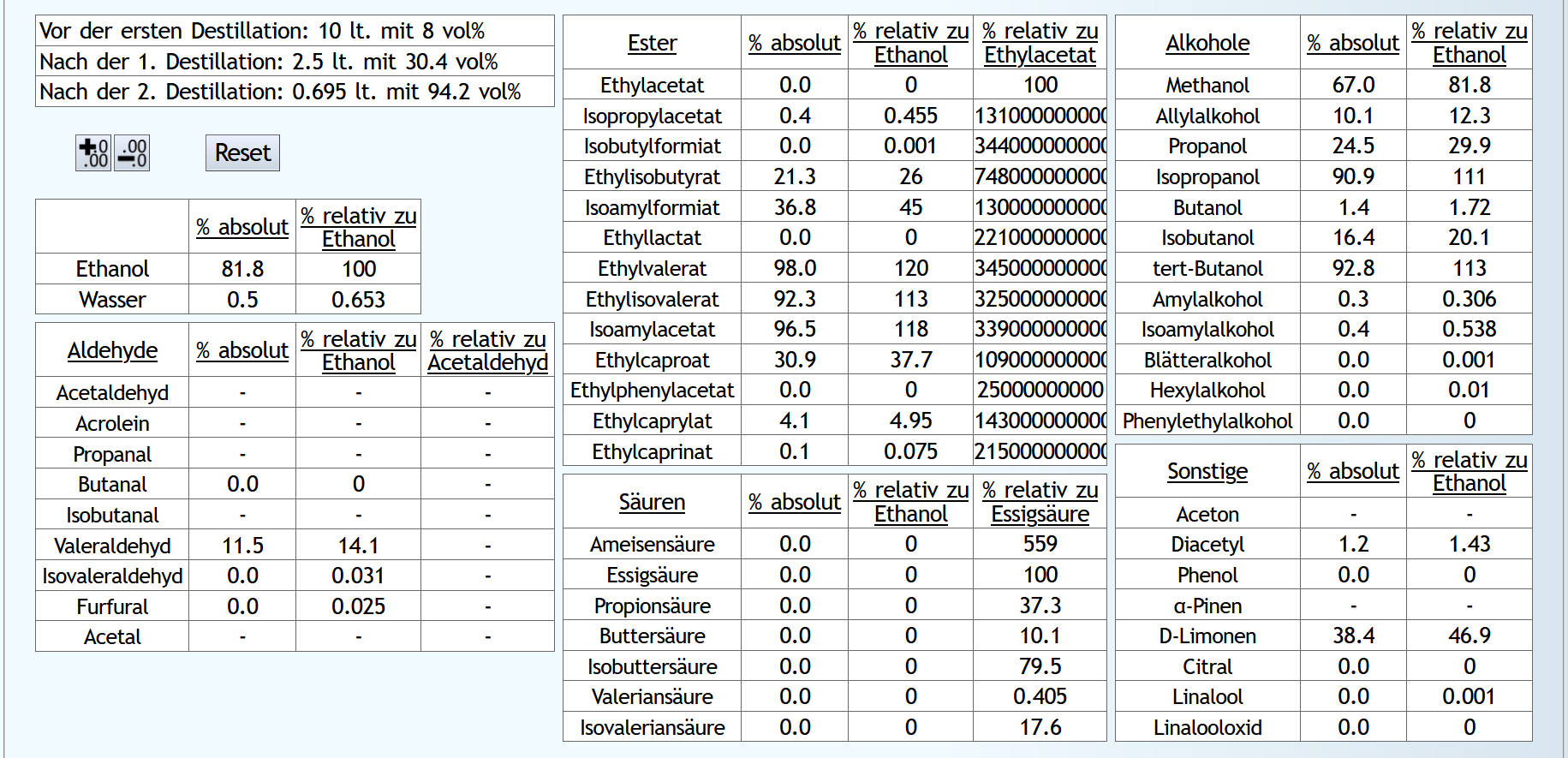 B)
B)
Raubrand: bis 2.5lt mit 1.1 theor. Böden. Dann 2.5lt Wasser dazu.
Feinbrand: 0.03lt Vorlauf mit 6 theor. Böden. Dann Mittellauf bis 90% des Alkohols destilliert ist; ebenfalls mit 6 theor. Böden.
Das Rechenergebnis für den Mittellauf: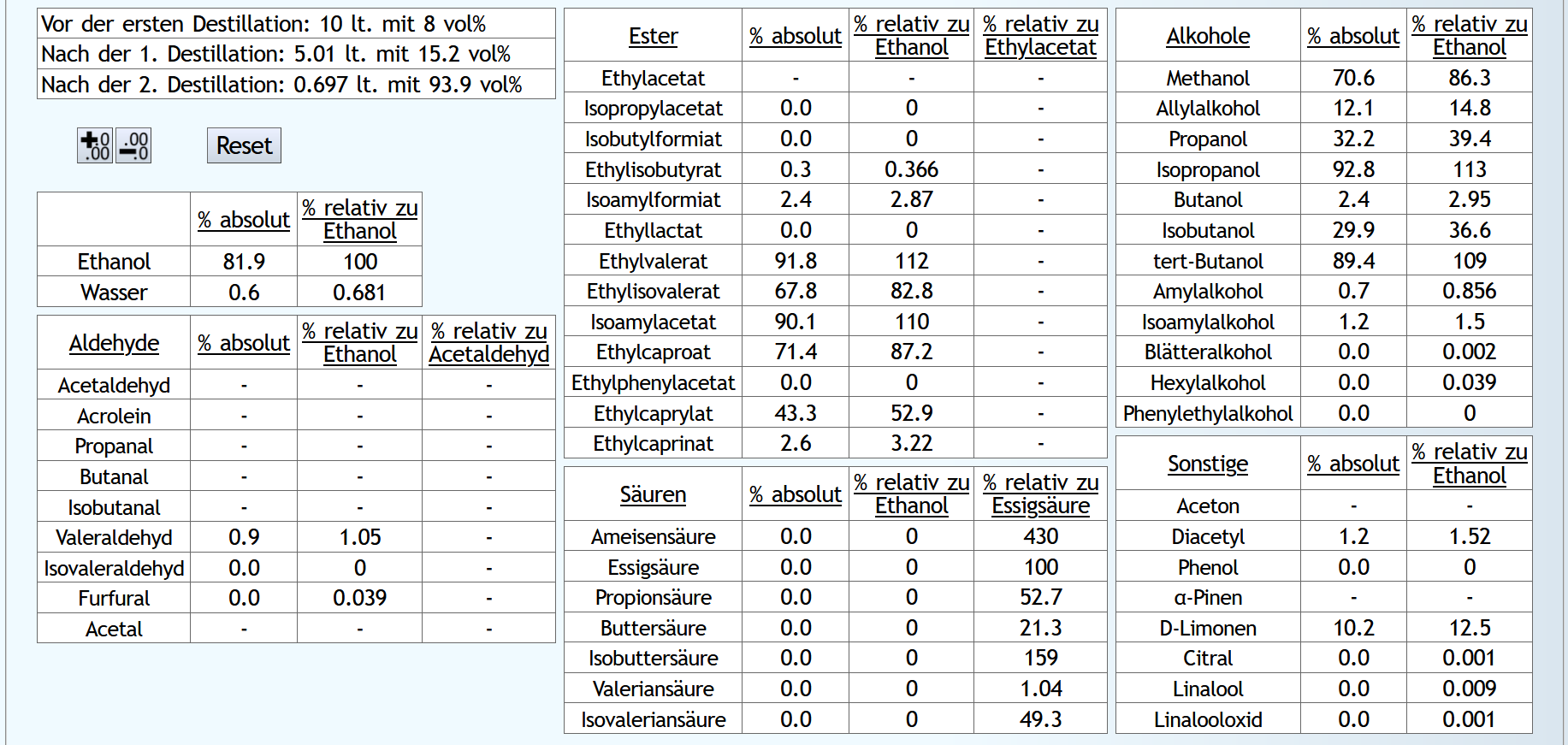 Mit Wasser sind die Aldehyde etwas besser abgetrennt.
Mit Wasser sind wesentlich weniger niedrige Ester im Mittellauf, aber teilweise mehr von den besseren.
Hier zeigt sich, daß der sehr kleine Vorlaufschnitt nicht ausreicht.
Ohne Wasser sind die höheren Alkohole etwas besser unterdrückt.
Aber allgemein sieht man hier, daß 6 theor. Böden nicht ausreichen, um die höheren Alkohole abzutrennen.
Bei den Sonstigen fällt D-Limonen auf, welches durch die Verdünnung stark in den Vorlauf gedrängt wurde.
Aber wie bei den Estern benötigt es hier einfach einen größeren Vorlaufschnitt.
Mit Wasser sind die Aldehyde etwas besser abgetrennt.
Mit Wasser sind wesentlich weniger niedrige Ester im Mittellauf, aber teilweise mehr von den besseren.
Hier zeigt sich, daß der sehr kleine Vorlaufschnitt nicht ausreicht.
Ohne Wasser sind die höheren Alkohole etwas besser unterdrückt.
Aber allgemein sieht man hier, daß 6 theor. Böden nicht ausreichen, um die höheren Alkohole abzutrennen.
Bei den Sonstigen fällt D-Limonen auf, welches durch die Verdünnung stark in den Vorlauf gedrängt wurde.
Aber wie bei den Estern benötigt es hier einfach einen größeren Vorlaufschnitt.
Fazit: Verdünnen ist vom Geruch her neutraler und besser. Die Destillation kostet aber etwas mehr Energie und natürlich einen größeren Kessel für die gleiche Menge an Alkohol. Man kann also überlegen, bei Neutral-Feinbränden unnötigen Freiraum im Kessel immer mit Wasser aufzufüllen.
10lt mit 8vol%.
Um ein deutliches Ergebnis zu bekommen, wird nur wenig Vorlauf abgetrennt, nicht allzustark rektifiziert und recht spät der Mittellauf beendet.
A)
Raubrand: bis 2.5lt mit 1.1 theor. Böden.
Feinbrand: 0.03lt Vorlauf mit 6 theor. Böden. Dann Mittellauf bis 90% des Alkohols destilliert ist; ebenfalls mit 6 theor. Böden.
Das Rechenergebnis für den Mittellauf:
Raubrand: bis 2.5lt mit 1.1 theor. Böden. Dann 2.5lt Wasser dazu.
Feinbrand: 0.03lt Vorlauf mit 6 theor. Böden. Dann Mittellauf bis 90% des Alkohols destilliert ist; ebenfalls mit 6 theor. Böden.
Das Rechenergebnis für den Mittellauf:
Fazit: Verdünnen ist vom Geruch her neutraler und besser. Die Destillation kostet aber etwas mehr Energie und natürlich einen größeren Kessel für die gleiche Menge an Alkohol. Man kann also überlegen, bei Neutral-Feinbränden unnötigen Freiraum im Kessel immer mit Wasser aufzufüllen.
Turbo-Einfachbrand vs Turbo-Doppelbrand
Hochgradige Maischen kann man theoretisch mit nur einer einzigen Potstill-Destillation auf eine gute Alkoholstärke destillieren.
Eine Doppeldestillation ist dafür eigentlich nicht unbedingt nötig.
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 2 berechnet.
10l mit 19vol%:
A) Einfachbrand:
Vorlauf 0.1lt mit 1.5 theor. Böden, dann Mittellauf bis 91°C im Dampf mit 1.2 theor. Böden. Bis 91°C, weil das in der Literatur der am häufigsten genannte Abtrennpunkt für Turbo-Einfachbrände ist. Die Daten des Mittellaufs: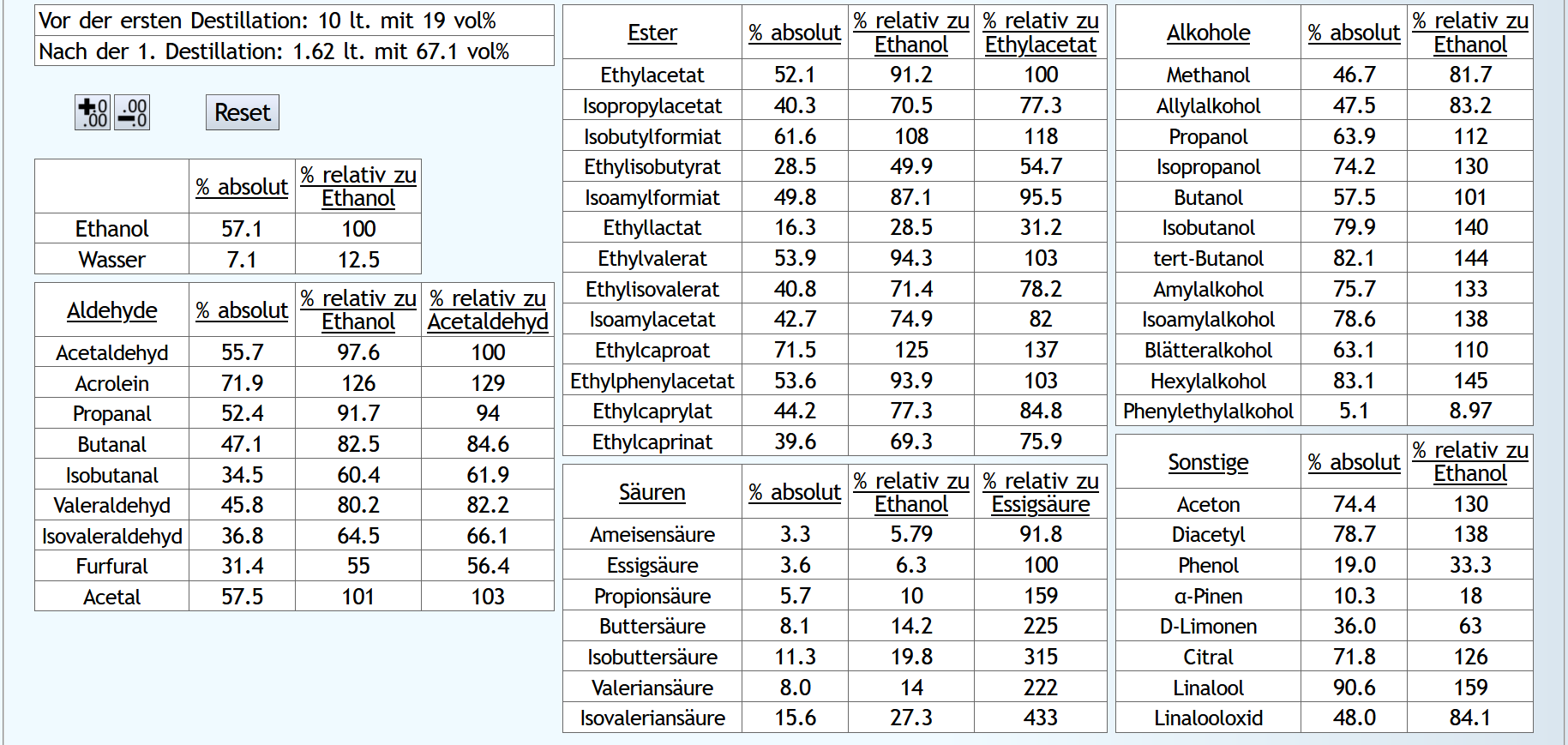 B) Doppelbrand:
B) Doppelbrand:
Raubrand bis 3.5lt mit 1.1 theor. Böden.
Feinbrand, Vorlauf 0.08lt mit 1.5 theor. Böden, dann Mittellauf bis aktuell 74vol% mit 1.2 theor. Böden. Die Daten des Mittellaufs: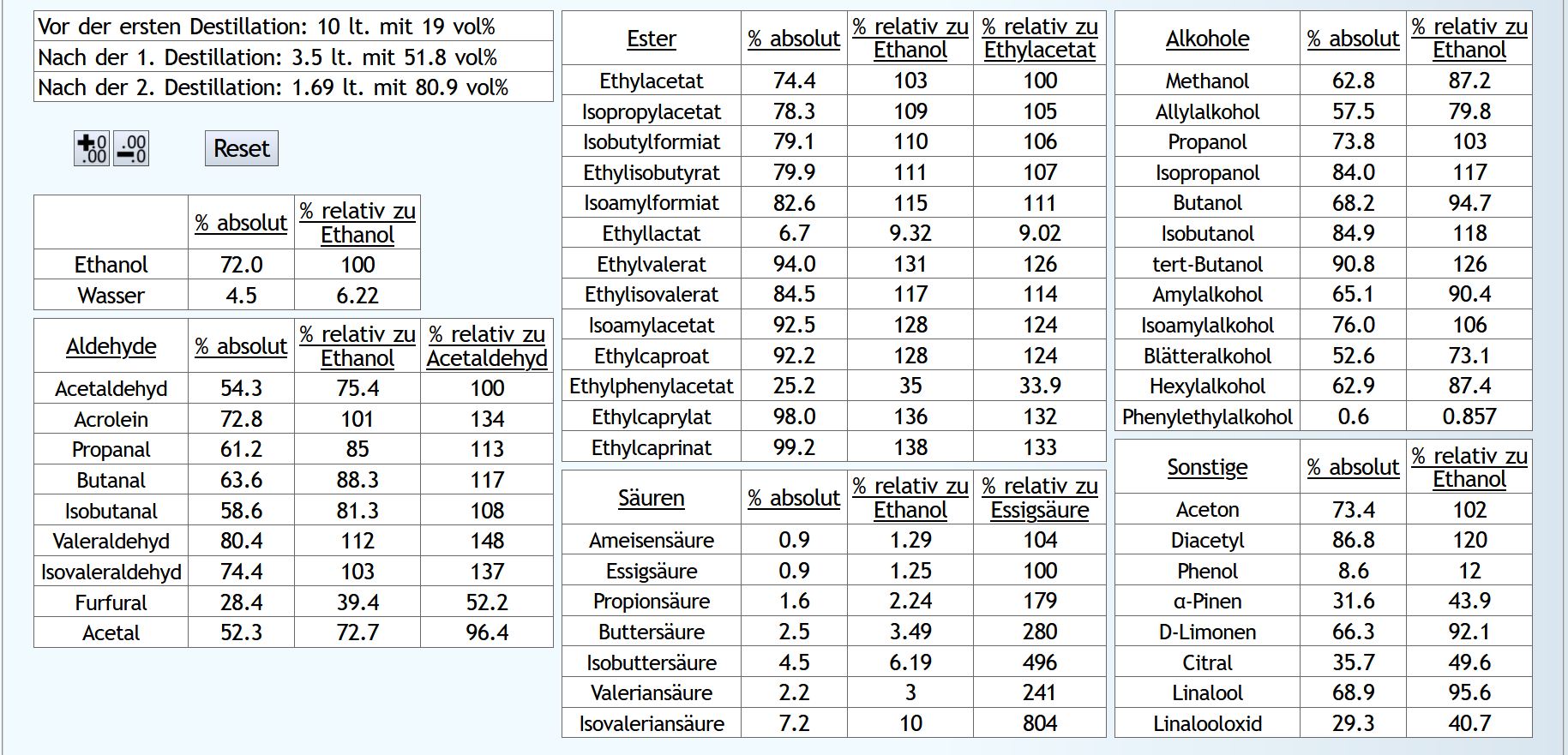 Der Doppelbrand hat 72% anstelle 57% Ethanolausbeute.
Der Doppelbrand hat etwas mehr Aldehde, und zwar von den guten.
Im Verhältnis zu seiner hohen Ethanolausbeute sind es allerdings insgesamt etwas weniger Aldehyde.
Bei den Estern ist es ähnlich.
Der Doppelbrand ist insgesamt etwas esterlastiger und dafür aldehydärmer.
Der Einfachbrand hat sehr viel mehr Säuren, ist also wesentlich kratziger auf der Zunge.
Der Doppelbrand hat mehr höhere Alkohole, im Verhältnis zur höheren Ethanolausbeute aber etwas weniger.
Der Doppelbrand hat 72% anstelle 57% Ethanolausbeute.
Der Doppelbrand hat etwas mehr Aldehde, und zwar von den guten.
Im Verhältnis zu seiner hohen Ethanolausbeute sind es allerdings insgesamt etwas weniger Aldehyde.
Bei den Estern ist es ähnlich.
Der Doppelbrand ist insgesamt etwas esterlastiger und dafür aldehydärmer.
Der Einfachbrand hat sehr viel mehr Säuren, ist also wesentlich kratziger auf der Zunge.
Der Doppelbrand hat mehr höhere Alkohole, im Verhältnis zur höheren Ethanolausbeute aber etwas weniger.
Fazit: Durch die sehr unterschiedliche Ethanolausbeute sind die beiden Brände sehr schlecht vergleichbar. Dadurch, daß man beim Einfachbrand so viel Ethanol verwirft, verteilen sich die flüchtigen Aromen auf weniger Alkohol. Allerdings ist sein Säuregehalt sehr hoch, was ihn kratzig macht. Außerdem ist das Verhältnis von guten zu schlechten Stoffen innerhalb einer Stoffgruppe beim Doppelbrand meist besser.
Wenn man, um die Ausbeute beim Einfachbrand zu erhöhen, den den Mittellauf später beendet, verliert man den Vorteil des Einfachbrands, daß sich die Aromen auf weniger Alkohol verteilen, und es kommen immer mehr Säuren. Seine niedrige Ausbeute rettet also den Einfachbrand. Ohne das Opfern dieser hohen Menge an Alkohol verliert man Qualität.
Nun könnte man beim Doppelbrand meinen, daß man beim Feinbrand nicht so hohe vol% im Kessel haben sollte. Dafür muss man entweder zwischen den Bränden verdünnen oder den Raubrand verlängern.
C) Doppelbrand wie B, aber zwischen den Bränden 1lt Wasser dazu und beim Feinbrand wegen des niedrigeren Alkoholgehalts im Kessel erst bei 68vol% aktuellem Alkoholgehalt den Mittellauf beendet. Die Daten des Mittellaufs: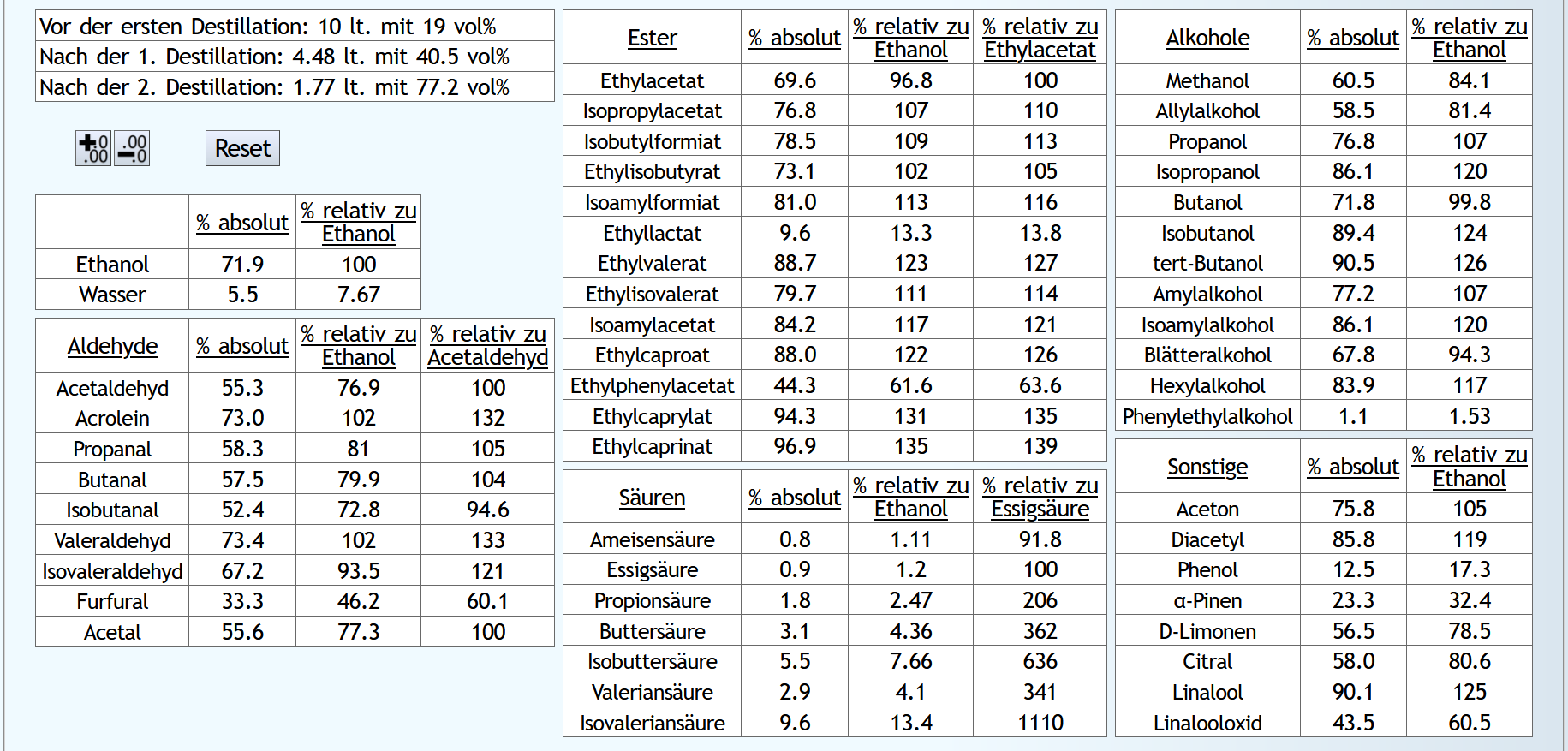 Das ist bei den Aldehyden und Alkoholen etwas schlechter und bei den Estern etwas besser.
Und hat eine etwas geringere Ethanolausbeute.
Bei den Säuren schaut es schlechter aus, aber bei der wichtigsten Säure, der Essigsäure, ist es sogar etwas besser.
Das ist bei den Aldehyden und Alkoholen etwas schlechter und bei den Estern etwas besser.
Und hat eine etwas geringere Ethanolausbeute.
Bei den Säuren schaut es schlechter aus, aber bei der wichtigsten Säure, der Essigsäure, ist es sogar etwas besser.
D) Doppelbrand wie C, aber anstelle 1lt Wasser dazuzugeben wird 1lt mehr raugebrannt und der Mittellauf wegen des etwas höheren Alkoholgehalts im Kessel erst bei 70vol% aktuellem Alkoholgehalt beendet. Die Daten des Mittellaufs: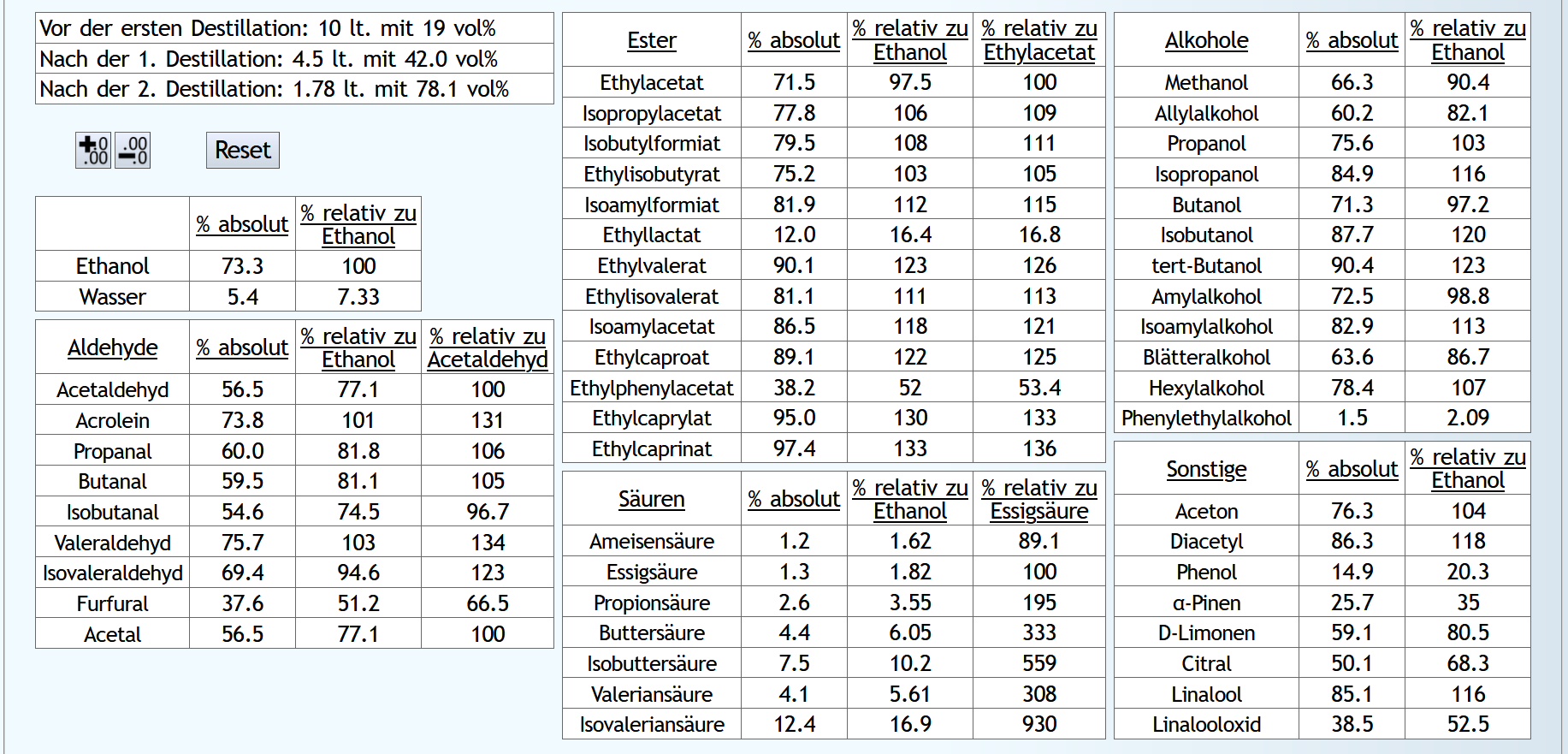 D hat natürlich mehr Begleitstoffe als C.
Bei den typischen Vorlaufstoffen ist das kaum merkbar.
Der verlängerte Raubrand von D bringt aber viel mehr Säuren.
Deswegen wird er weniger weich schmecken als der mit Wasser verdünnte C.
D hat natürlich mehr Begleitstoffe als C.
Bei den typischen Vorlaufstoffen ist das kaum merkbar.
Der verlängerte Raubrand von D bringt aber viel mehr Säuren.
Deswegen wird er weniger weich schmecken als der mit Wasser verdünnte C.
Die beiden besten Versionen B und C sind recht ähnlich.
Gesamtfazit: Der Einfachbrand schaut bei den Säuren und bei der Mittellaufausbeute sehr schlecht aus. Aber die schlechte Ethanolausbeute rettet ihn in gewisser Weise. Erhöht man diese, wird er sauer. Beim Doppelbrand ist das extrem weit herunterbrennen des Raubrands, um beim Feinbrand nicht allzu hohe vol% im Kessel zu haben, eine schlechte Empfehlung. Weniger weit herunterzubrennen und dann entweder verdünnt oder unverdünnt zu brennen, ist beides besser.
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 2 berechnet.
10l mit 19vol%:
A) Einfachbrand:
Vorlauf 0.1lt mit 1.5 theor. Böden, dann Mittellauf bis 91°C im Dampf mit 1.2 theor. Böden. Bis 91°C, weil das in der Literatur der am häufigsten genannte Abtrennpunkt für Turbo-Einfachbrände ist. Die Daten des Mittellaufs:
Raubrand bis 3.5lt mit 1.1 theor. Böden.
Feinbrand, Vorlauf 0.08lt mit 1.5 theor. Böden, dann Mittellauf bis aktuell 74vol% mit 1.2 theor. Böden. Die Daten des Mittellaufs:
Fazit: Durch die sehr unterschiedliche Ethanolausbeute sind die beiden Brände sehr schlecht vergleichbar. Dadurch, daß man beim Einfachbrand so viel Ethanol verwirft, verteilen sich die flüchtigen Aromen auf weniger Alkohol. Allerdings ist sein Säuregehalt sehr hoch, was ihn kratzig macht. Außerdem ist das Verhältnis von guten zu schlechten Stoffen innerhalb einer Stoffgruppe beim Doppelbrand meist besser.
Wenn man, um die Ausbeute beim Einfachbrand zu erhöhen, den den Mittellauf später beendet, verliert man den Vorteil des Einfachbrands, daß sich die Aromen auf weniger Alkohol verteilen, und es kommen immer mehr Säuren. Seine niedrige Ausbeute rettet also den Einfachbrand. Ohne das Opfern dieser hohen Menge an Alkohol verliert man Qualität.
Nun könnte man beim Doppelbrand meinen, daß man beim Feinbrand nicht so hohe vol% im Kessel haben sollte. Dafür muss man entweder zwischen den Bränden verdünnen oder den Raubrand verlängern.
C) Doppelbrand wie B, aber zwischen den Bränden 1lt Wasser dazu und beim Feinbrand wegen des niedrigeren Alkoholgehalts im Kessel erst bei 68vol% aktuellem Alkoholgehalt den Mittellauf beendet. Die Daten des Mittellaufs:
D) Doppelbrand wie C, aber anstelle 1lt Wasser dazuzugeben wird 1lt mehr raugebrannt und der Mittellauf wegen des etwas höheren Alkoholgehalts im Kessel erst bei 70vol% aktuellem Alkoholgehalt beendet. Die Daten des Mittellaufs:
Die beiden besten Versionen B und C sind recht ähnlich.
Gesamtfazit: Der Einfachbrand schaut bei den Säuren und bei der Mittellaufausbeute sehr schlecht aus. Aber die schlechte Ethanolausbeute rettet ihn in gewisser Weise. Erhöht man diese, wird er sauer. Beim Doppelbrand ist das extrem weit herunterbrennen des Raubrands, um beim Feinbrand nicht allzu hohe vol% im Kessel zu haben, eine schlechte Empfehlung. Weniger weit herunterzubrennen und dann entweder verdünnt oder unverdünnt zu brennen, ist beides besser.
Dreifach anstelle doppelt brennen?
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 2 berechnet.
Zuerst eine Maische mit sehr niedrigem Alkoholgehalt. 10lt Maische mit 5vol%:
A) Doppelbrand:
Raubrand: 2lt mit 1.1theor. Böden.
Feinbrand: 0.03lt Vorlauf mit 1.5 theor. Böden. Mittellauf mit 1.2 theor. Böden bis 55vol% aktuellem Alkoholgehalt.
Die Daten des Mittellaufs: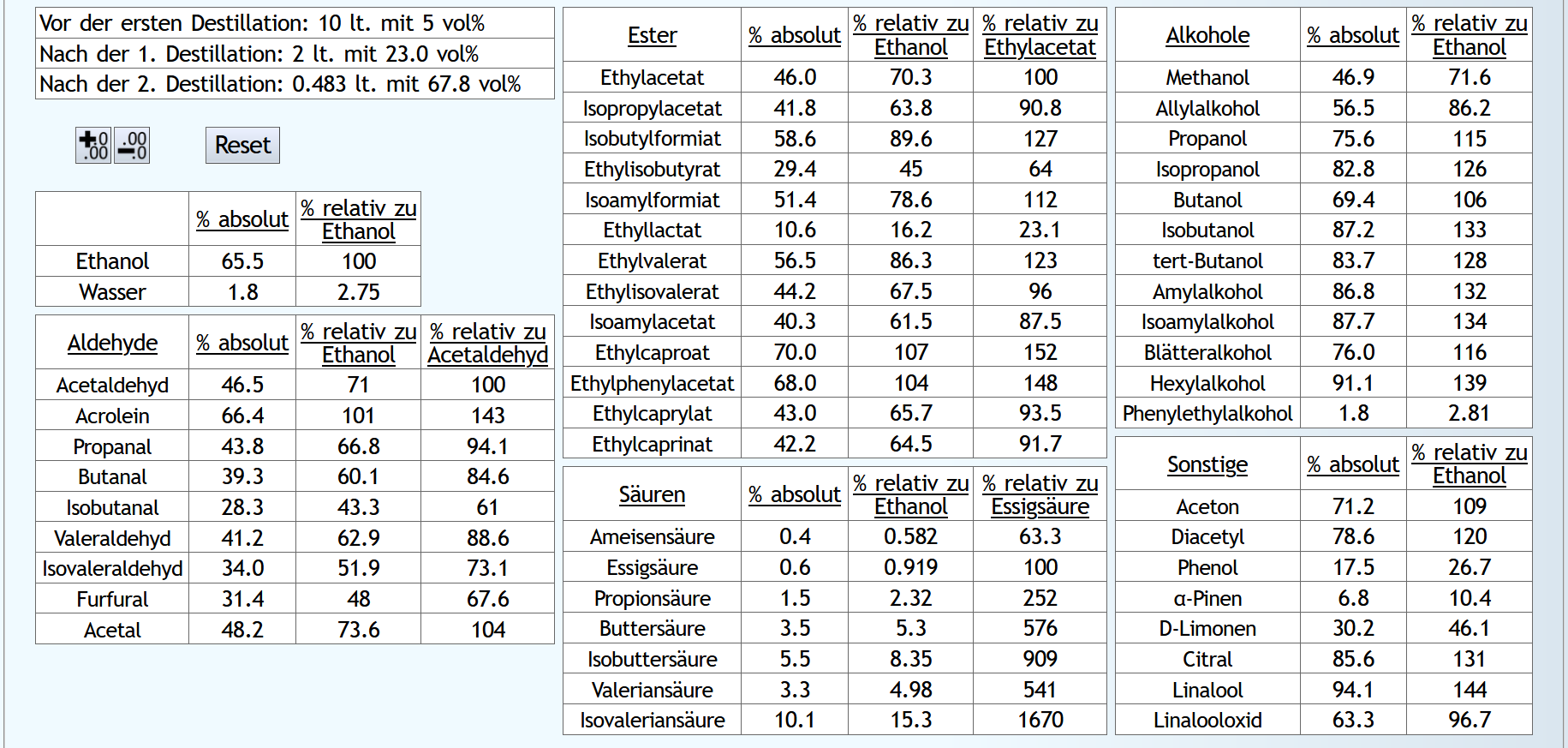 B) Dreifachbrand:
B) Dreifachbrand:
Raubrand: 3lt mit 1.1 theor. Böden.
Mittelbrand: 1.5lt mit 1.1 theor. Böden.
Feinbrand: 0.03lt Vorlauf mit 1.5 theor. Böden. Mittellauf mit 1.2 theor. Böden bis 65vol% aktuellem Alkoholgehalt.
Die Daten des Mittellaufs: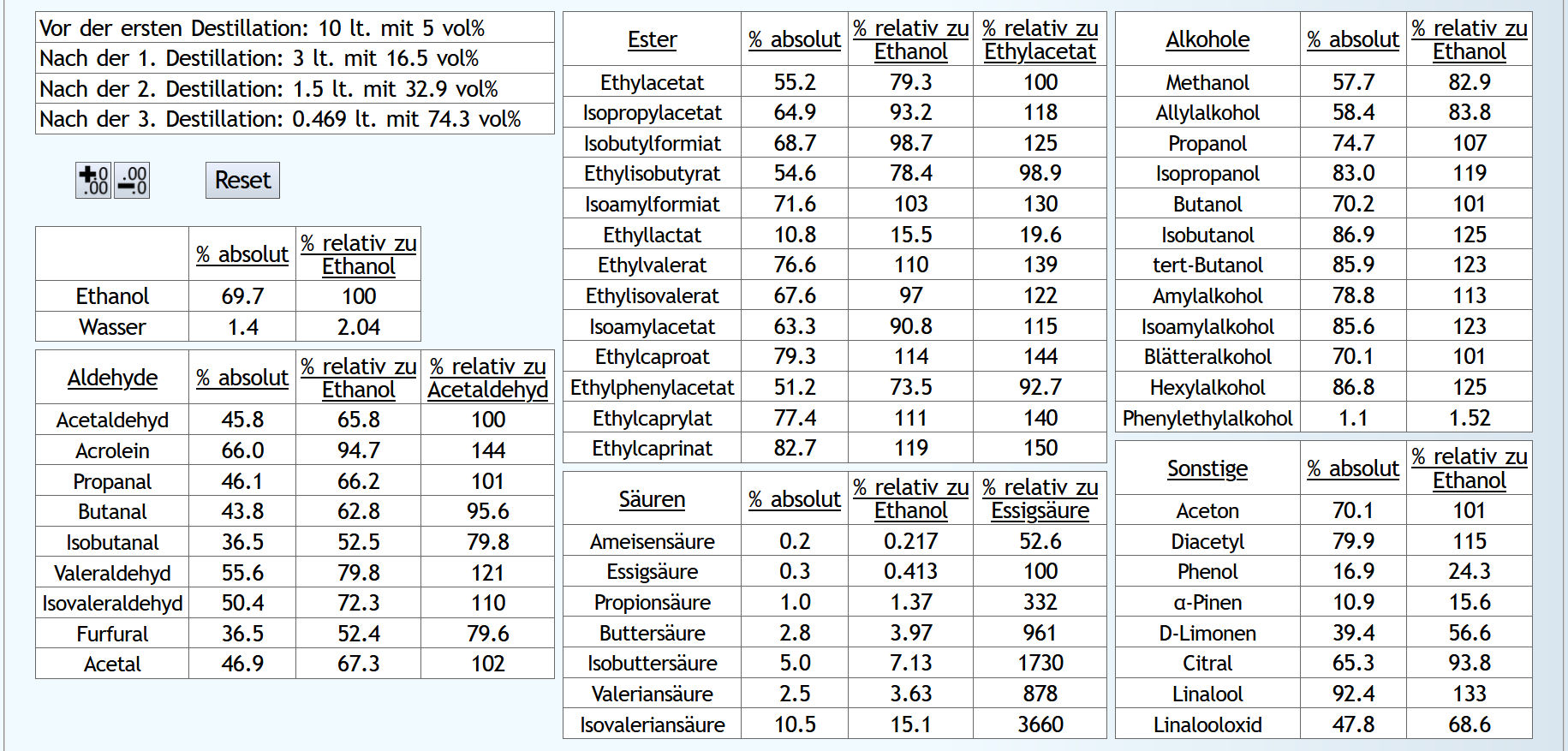 Die Ethanolausbeute ist beim Dreifachbrand etwas höher, 70 anstelle 65%.
Die Vorlaufabtrennung ist beim Doppelbrand etwas effektiver;
es wurden mehr Aldehyde und Ester abgetrennt.
Allerdings vor allem von den guten.
Höhere Alkohole sind minimal weniger im Doppelbrand.
Beim Dreifachbrand wurden deutlich mehr Säuren abgetrennt.
Bei den Sonstigen Stoffen ist es unterschiedlich.
Die Ethanolausbeute ist beim Dreifachbrand etwas höher, 70 anstelle 65%.
Die Vorlaufabtrennung ist beim Doppelbrand etwas effektiver;
es wurden mehr Aldehyde und Ester abgetrennt.
Allerdings vor allem von den guten.
Höhere Alkohole sind minimal weniger im Doppelbrand.
Beim Dreifachbrand wurden deutlich mehr Säuren abgetrennt.
Bei den Sonstigen Stoffen ist es unterschiedlich.
Wegen den reduzierten Säuren und dem besseren Verhältnis von guten zu schlechten Aldehyden bzw. guten zu schlechten Ester ist der Dreifachbrand also besser.
Ist das aber bei höherprozentiger Maische auch so?
10lt Maische mit 12vol%:
C) Doppelbrand:
Raubrand: 3.3lt mit 1.1 theor. Böden.
Feinbrand: 0.05lt Vorlauf mit 1.5 theor. Böden. Mittellauf mit 1.2 theor. Böden bis 66vol% aktuellem Alkoholgehalt.
Die Daten des Mittellaufs: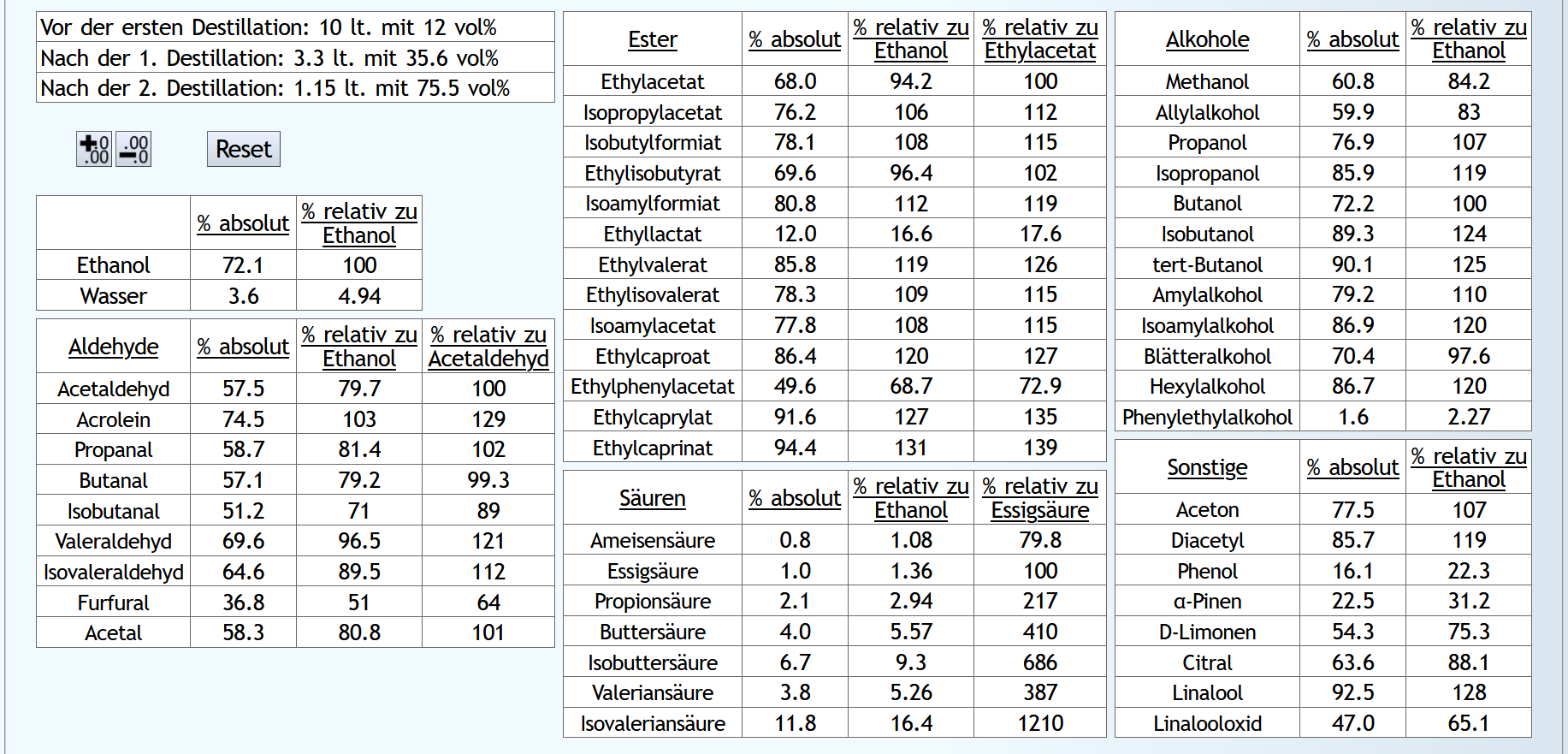 D) Dreifachbrand:
D) Dreifachbrand:
Raubrand: 4lt mit 1.1 theor. Böden.
Mittelbrand: 2.5lt mit 1.1 theor. Böden.
Feinbrand: Vorlauf 0.05lt mit 1.5 theor. Böden. Mittellauf mit 1.2 theor. Böden bis 72vol% aktuellem Alkoholgehalt.
Die Daten des Mittellaufs: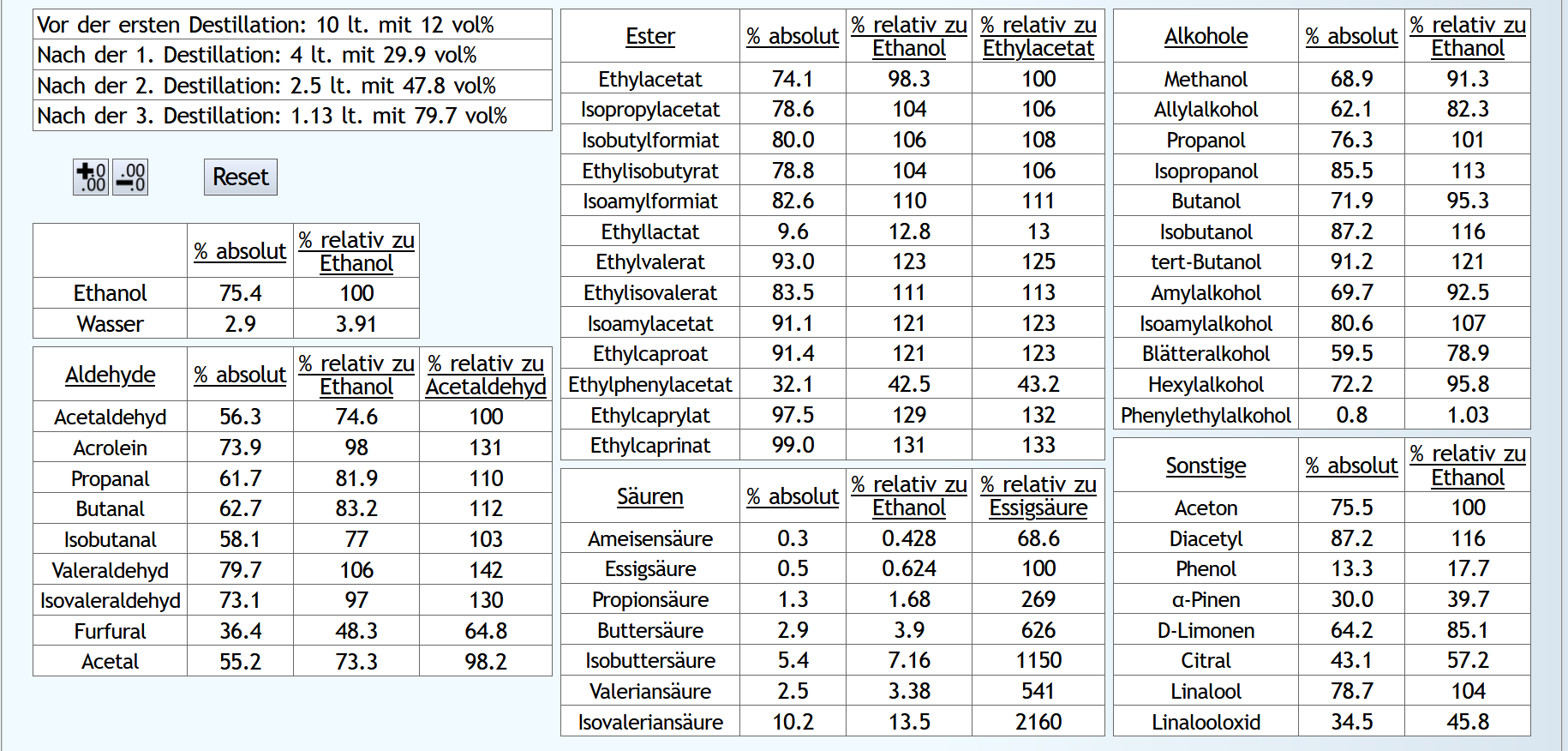 Die Ergebnisse sind ähnlich wie bei niedrigem Alkoholgehalt.
Bei den Estern ist der Vorteil des Dreifachbrands größtenteils verschwunden.
Bei den Säuren ist der Unterschied vielleicht noch etwas größer geworden.
Der Dreifachbrand schaut also auch bei höherprozentigen Maischen besser als der Doppelbrand aus.
Aber es ist nicht mehr so leicht ersichtlicht.
Die Ergebnisse sind ähnlich wie bei niedrigem Alkoholgehalt.
Bei den Estern ist der Vorteil des Dreifachbrands größtenteils verschwunden.
Bei den Säuren ist der Unterschied vielleicht noch etwas größer geworden.
Der Dreifachbrand schaut also auch bei höherprozentigen Maischen besser als der Doppelbrand aus.
Aber es ist nicht mehr so leicht ersichtlicht.
Ob der Doppelbrand irgendwann im Vorteil ist, wenn man die vol% in der Maische weiter erhöht?
10lt Maische mit 19vol%:
E) Doppelbrand:
Raubrand: 3.5lt mit 1.1 theor. Böden.
Feinbrand: Vorlauf 0.08lt mit 1.5 theor. Böden. Mittellauf mit 1.2 theor. Böden bis 72vol% aktuellem Alkoholgehalt.
Die Daten des Mittellaufs: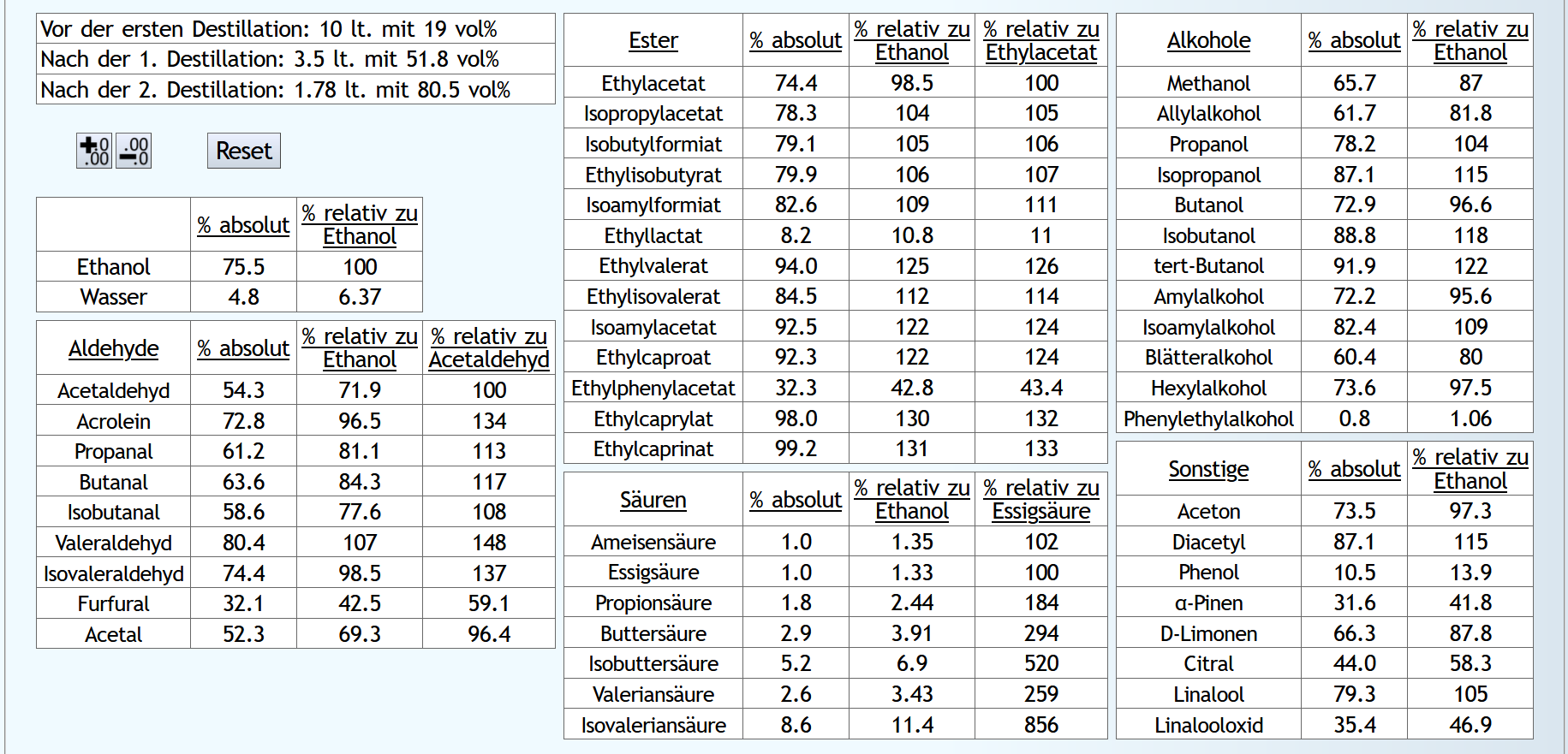 F) Dreifachbrand:
F) Dreifachbrand:
Raubrand: 4lt mit 1.1 theor. Böden.
Mittelbrand: 3lt mit 1.1 theor. Böden.
Feinbrand: Vorlauf 0.08lt mit 1.5 theor. Böden. Mittellauf mit 1.2 theor. Böden bis 78vol% aktuellem Alkoholgehalt.
Die Daten des Mittellaufs: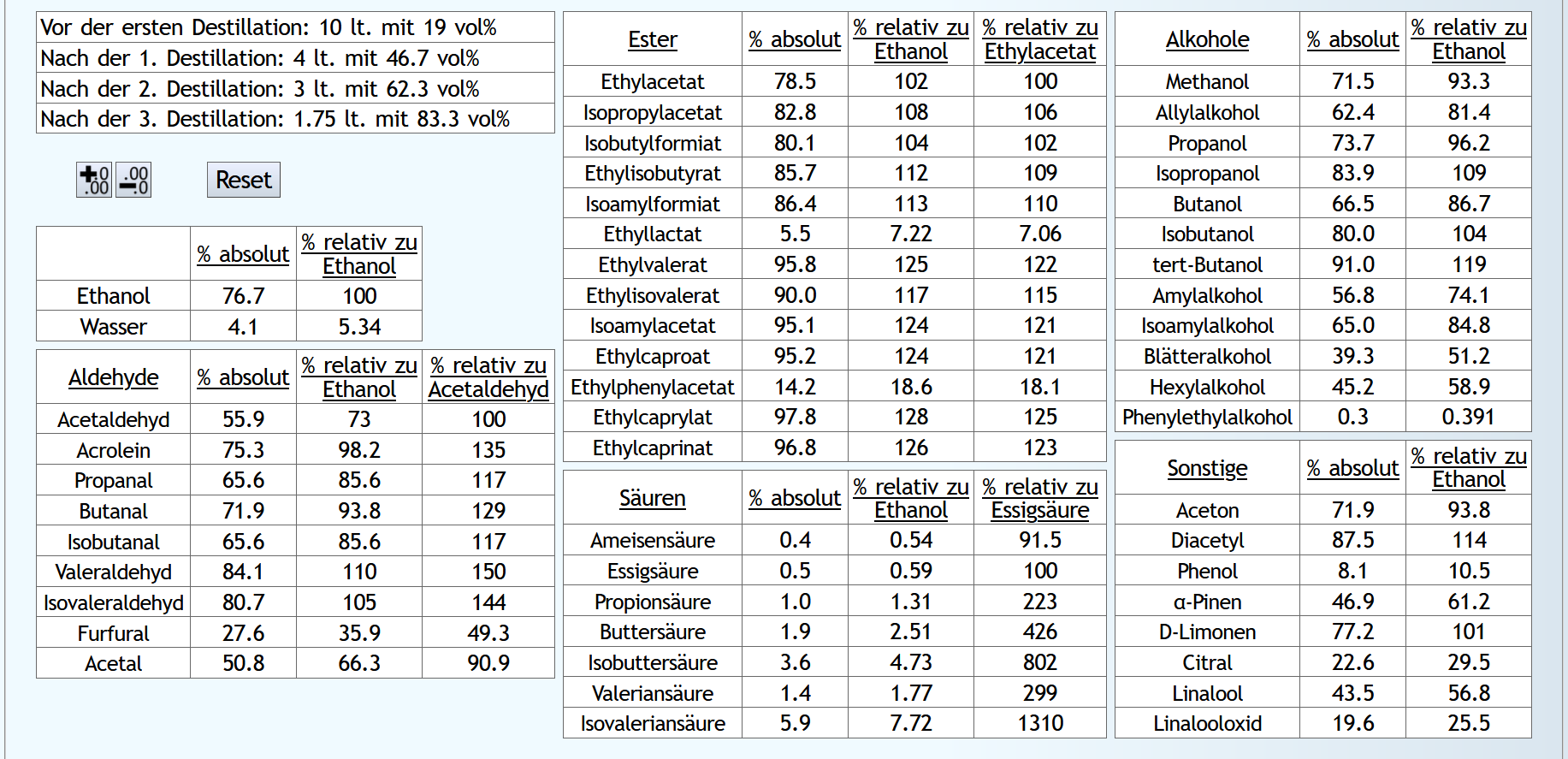 Der Dreifachbrand hat nur noch einen minimalen Vorteil bei den Aldehyden.
Bei den Estern beginnt er, qualitativ schlechter zu werden.
Bei der Abtrennung der höheren Alkohole wird er besser als der Doppelbrand.
Bei den Säuren ist er immer noch viel besser.
Der Dreifachbrand hat nur noch einen minimalen Vorteil bei den Aldehyden.
Bei den Estern beginnt er, qualitativ schlechter zu werden.
Bei der Abtrennung der höheren Alkohole wird er besser als der Doppelbrand.
Bei den Säuren ist er immer noch viel besser.
Fazit: Es schaut so aus, als wenn der Dreifachbrand meistens besser ist. Allerdings schmilzt sein Vorteil, je alkoholstärker die Maische ist. Bei sehr niedrigprozentiger Maische ist er klar im Vorteil, bei Turbomaische ist es wohl auch Geschmackssache.
Zuerst eine Maische mit sehr niedrigem Alkoholgehalt. 10lt Maische mit 5vol%:
A) Doppelbrand:
Raubrand: 2lt mit 1.1theor. Böden.
Feinbrand: 0.03lt Vorlauf mit 1.5 theor. Böden. Mittellauf mit 1.2 theor. Böden bis 55vol% aktuellem Alkoholgehalt.
Die Daten des Mittellaufs:
Raubrand: 3lt mit 1.1 theor. Böden.
Mittelbrand: 1.5lt mit 1.1 theor. Böden.
Feinbrand: 0.03lt Vorlauf mit 1.5 theor. Böden. Mittellauf mit 1.2 theor. Böden bis 65vol% aktuellem Alkoholgehalt.
Die Daten des Mittellaufs:
Wegen den reduzierten Säuren und dem besseren Verhältnis von guten zu schlechten Aldehyden bzw. guten zu schlechten Ester ist der Dreifachbrand also besser.
Ist das aber bei höherprozentiger Maische auch so?
10lt Maische mit 12vol%:
C) Doppelbrand:
Raubrand: 3.3lt mit 1.1 theor. Böden.
Feinbrand: 0.05lt Vorlauf mit 1.5 theor. Böden. Mittellauf mit 1.2 theor. Böden bis 66vol% aktuellem Alkoholgehalt.
Die Daten des Mittellaufs:
Raubrand: 4lt mit 1.1 theor. Böden.
Mittelbrand: 2.5lt mit 1.1 theor. Böden.
Feinbrand: Vorlauf 0.05lt mit 1.5 theor. Böden. Mittellauf mit 1.2 theor. Böden bis 72vol% aktuellem Alkoholgehalt.
Die Daten des Mittellaufs:
Ob der Doppelbrand irgendwann im Vorteil ist, wenn man die vol% in der Maische weiter erhöht?
10lt Maische mit 19vol%:
E) Doppelbrand:
Raubrand: 3.5lt mit 1.1 theor. Böden.
Feinbrand: Vorlauf 0.08lt mit 1.5 theor. Böden. Mittellauf mit 1.2 theor. Böden bis 72vol% aktuellem Alkoholgehalt.
Die Daten des Mittellaufs:
Raubrand: 4lt mit 1.1 theor. Böden.
Mittelbrand: 3lt mit 1.1 theor. Böden.
Feinbrand: Vorlauf 0.08lt mit 1.5 theor. Böden. Mittellauf mit 1.2 theor. Böden bis 78vol% aktuellem Alkoholgehalt.
Die Daten des Mittellaufs:
Fazit: Es schaut so aus, als wenn der Dreifachbrand meistens besser ist. Allerdings schmilzt sein Vorteil, je alkoholstärker die Maische ist. Bei sehr niedrigprozentiger Maische ist er klar im Vorteil, bei Turbomaische ist es wohl auch Geschmackssache.
Doppelt mit Potstill oder einfach mit Refluxdestille brennen?
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 2 berechnet.
Dieser Vergleich ist schwer, fair durchzuführen. Denn wenn man einen Einfachbrand mit einer Refluxdestille macht, wird man den Vorlauf wahrscheinlich mit viel Reflux abtrennen. Diese Option hat man mit einer normalen Potstill aber nicht. Für einen Vergleich ist es interessanter, wenn die Refluxdestille testweise zuerst mal auf diese Möglichkeit verzichtet, auch wenn das dann erst einmal nicht der Realität entspricht:
10lt Maische mit 10vol%:
A) Potstill-Doppelbrand:
Raubrand: 3lt mit 1.1 theor. Böden.
Feinbrand: Vorlauf 0.05lt mit 1.5 theor. Böden. Mittellauf bis 65vol% aktuellem Alkoholgehalt mit 1.2 theor. Böden.
Die Daten des Mittellaufs: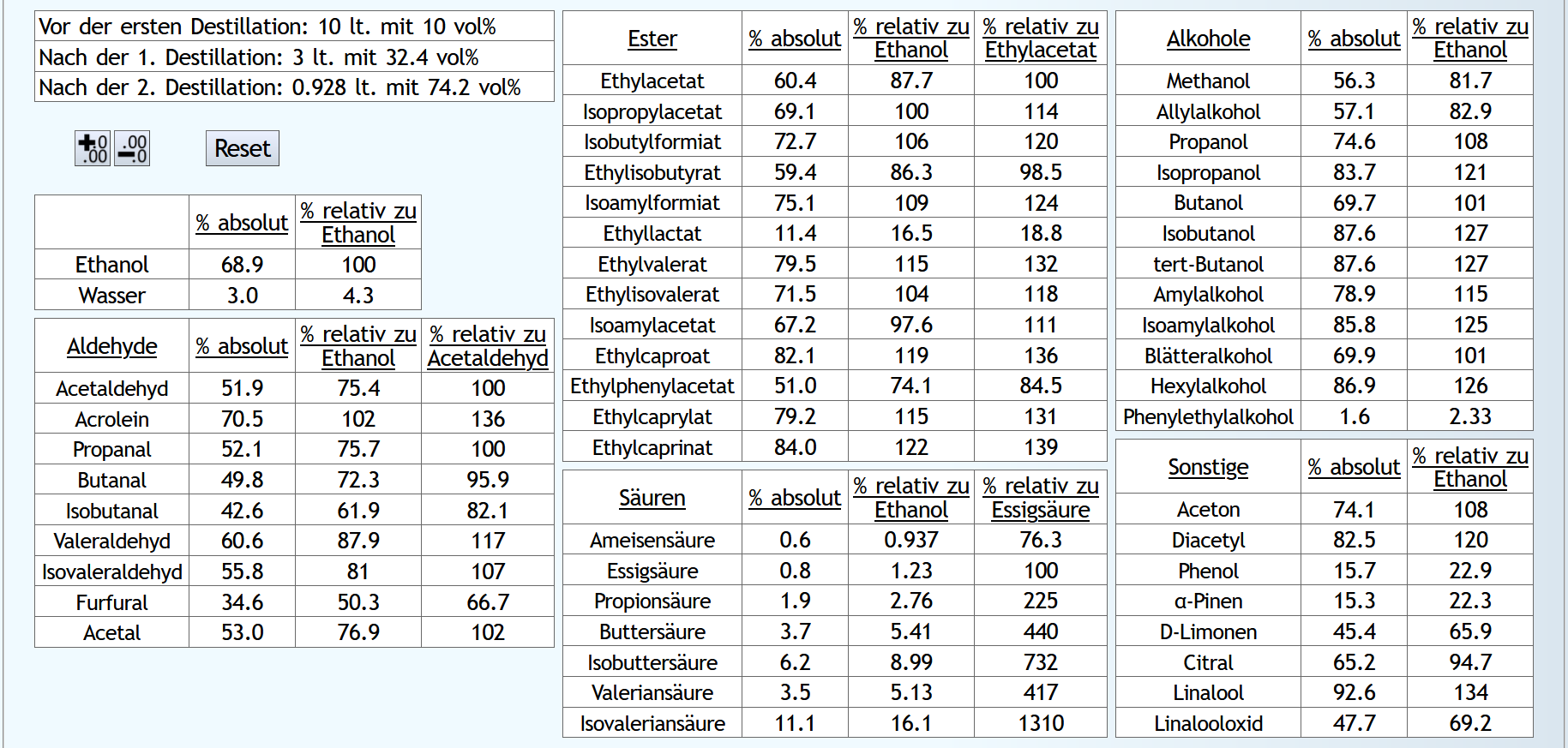 B) Reflux Einfachbrand:
B) Reflux Einfachbrand:
0.05lt Vorlauf mit 1.5 theor. Böden und dann versucht den Alkoholgehalt nicht unter 70vol% sinken zu lassen, bis der Mittellauf die gleiche Ausbeute hat wie die vom Potstill-Doppelbrand. Das ist das Protokoll: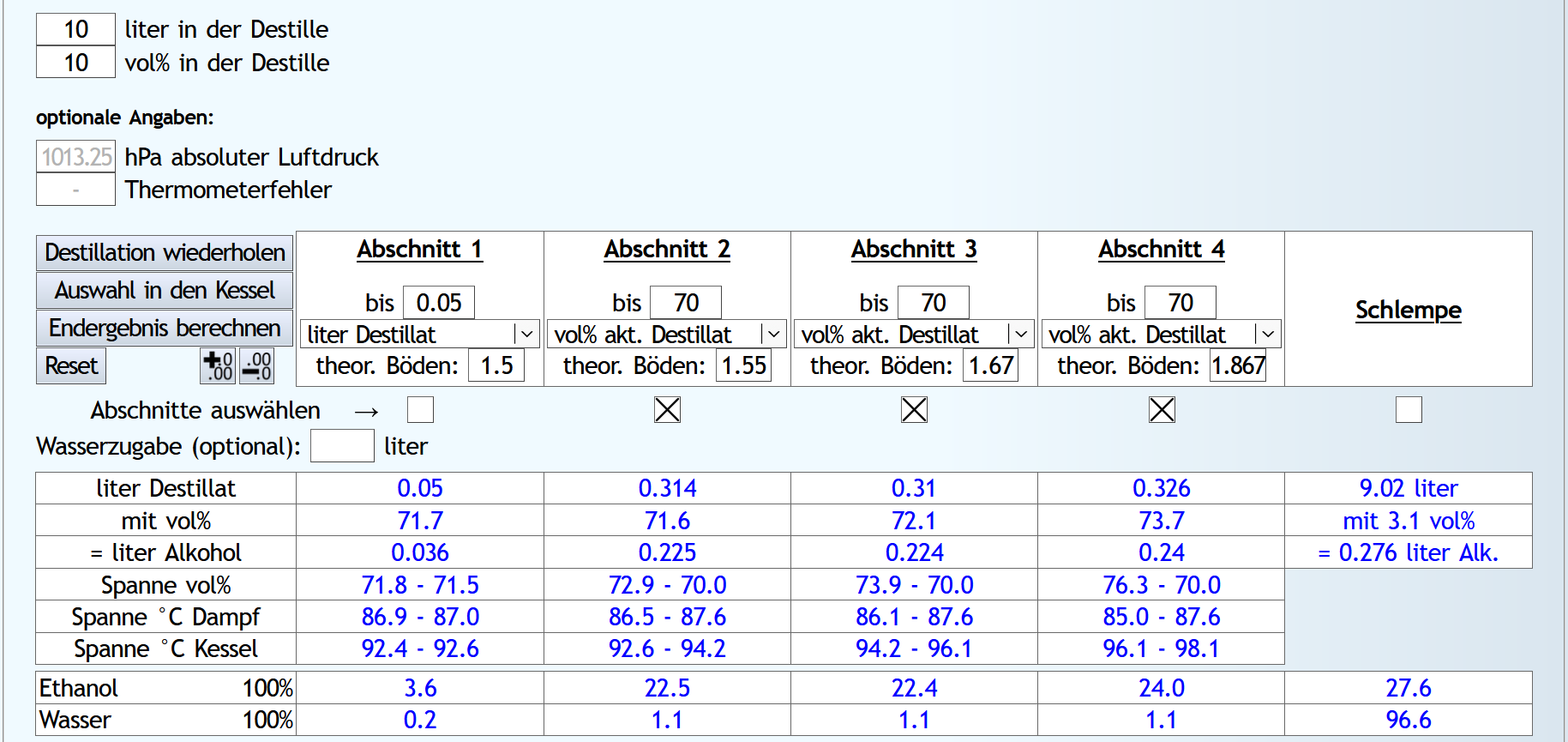 Und hier die Daten des Mittellaufs:
Und hier die Daten des Mittellaufs:
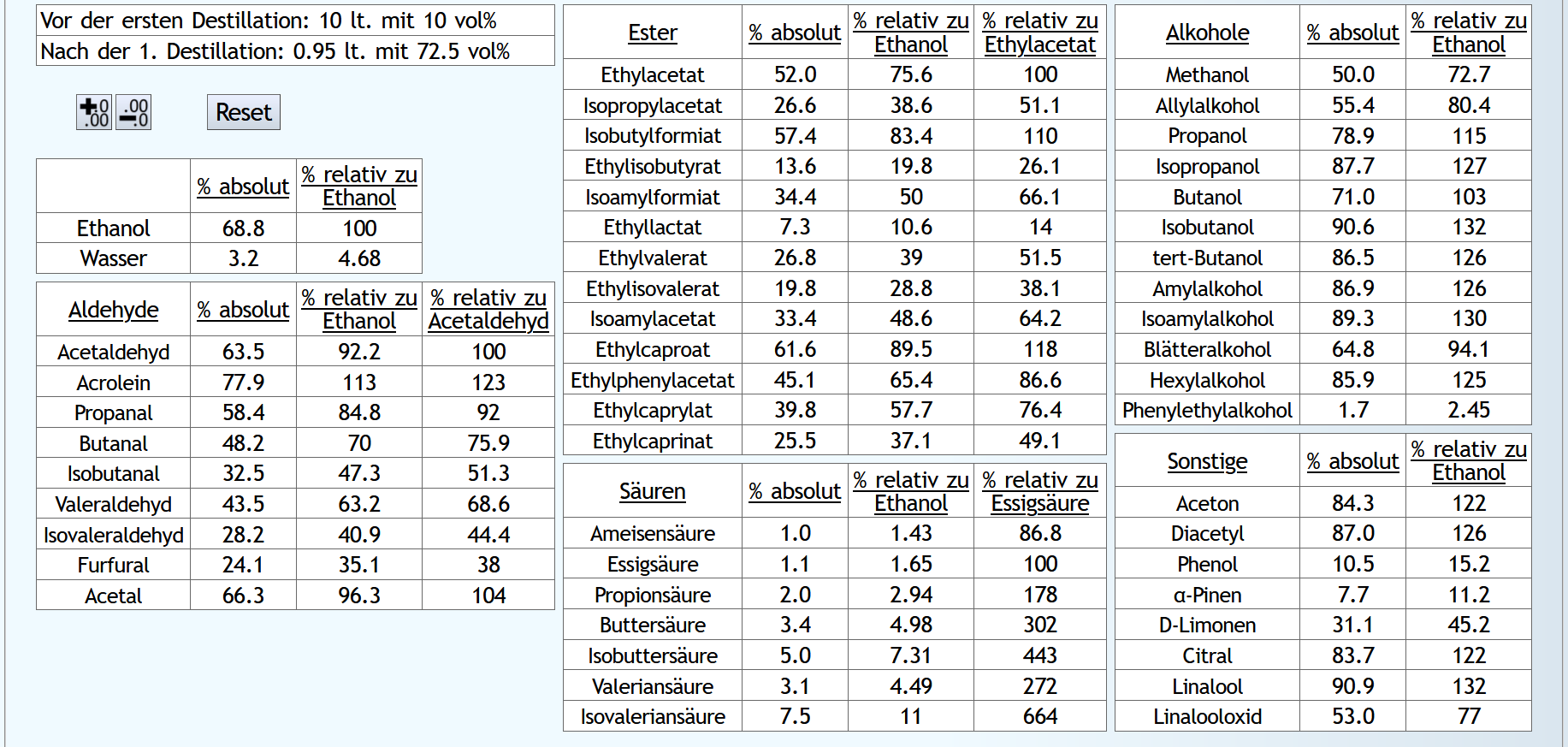 Der Doppelbrand hat mehr gute und weniger schlechte Aldehyde.
Er hat mehr Ester, auch von den schlechten, aber vor allem von den guten.
Er hat weniger Essigsäure, aber mehr von den größeren Säuren.
Da Essigsäure den Löwenanteil der Säuren ausmacht, ist er etwas weicher.
Der Doppelbrand hat also qualitative Vorzüge, braucht aber wahrscheinlich eine etwas großzügigere Vorlaufabtrennung, um mehr von den schlechten kleinen Aldehyden und Estern abzutrennen.
Der Doppelbrand hat mehr gute und weniger schlechte Aldehyde.
Er hat mehr Ester, auch von den schlechten, aber vor allem von den guten.
Er hat weniger Essigsäure, aber mehr von den größeren Säuren.
Da Essigsäure den Löwenanteil der Säuren ausmacht, ist er etwas weicher.
Der Doppelbrand hat also qualitative Vorzüge, braucht aber wahrscheinlich eine etwas großzügigere Vorlaufabtrennung, um mehr von den schlechten kleinen Aldehyden und Estern abzutrennen.
Aber dieser Vergleich war ja unfair, weil die Refluxdestille auf ihre Möglichkeit, den Vorlauf zu rektifizieren, verzichtet hat. Würde man den Vorlauf rektifizieren, würde er effizienter und gechmacklich besser abgetrennt werden. Also die schlechten Aldehyde und Ester würden mehr abgetrennt werden und die besseren weniger. Ein bisschen mehr höhere Alkohole hätte man allerdings im Mittellauf, da keine mehr mit dem Vorlauf abgetrennt werden, wenn man ihn rektifiziert. Und das Problem des Reflux-Einfachbrandes mit den Säuren würde sowieso bleiben.
Fazit: Das beste ist also die Mischung: eine Potstill-Doppeldestillation mit Rektifizierung des Vorlaufs.
Dieser Vergleich ist schwer, fair durchzuführen. Denn wenn man einen Einfachbrand mit einer Refluxdestille macht, wird man den Vorlauf wahrscheinlich mit viel Reflux abtrennen. Diese Option hat man mit einer normalen Potstill aber nicht. Für einen Vergleich ist es interessanter, wenn die Refluxdestille testweise zuerst mal auf diese Möglichkeit verzichtet, auch wenn das dann erst einmal nicht der Realität entspricht:
10lt Maische mit 10vol%:
A) Potstill-Doppelbrand:
Raubrand: 3lt mit 1.1 theor. Böden.
Feinbrand: Vorlauf 0.05lt mit 1.5 theor. Böden. Mittellauf bis 65vol% aktuellem Alkoholgehalt mit 1.2 theor. Böden.
Die Daten des Mittellaufs:
0.05lt Vorlauf mit 1.5 theor. Böden und dann versucht den Alkoholgehalt nicht unter 70vol% sinken zu lassen, bis der Mittellauf die gleiche Ausbeute hat wie die vom Potstill-Doppelbrand. Das ist das Protokoll:
Aber dieser Vergleich war ja unfair, weil die Refluxdestille auf ihre Möglichkeit, den Vorlauf zu rektifizieren, verzichtet hat. Würde man den Vorlauf rektifizieren, würde er effizienter und gechmacklich besser abgetrennt werden. Also die schlechten Aldehyde und Ester würden mehr abgetrennt werden und die besseren weniger. Ein bisschen mehr höhere Alkohole hätte man allerdings im Mittellauf, da keine mehr mit dem Vorlauf abgetrennt werden, wenn man ihn rektifiziert. Und das Problem des Reflux-Einfachbrandes mit den Säuren würde sowieso bleiben.
Fazit: Das beste ist also die Mischung: eine Potstill-Doppeldestillation mit Rektifizierung des Vorlaufs.
Dreifachbrand ohne Rektifikation oder Doppelbrand mit Rektifikation beim Raubrand?
Wenn der Doppelbrand mit Rektifikation beim Raubrand genausogut ist wie der Dreifachbrand ohne Rektifikation, könnte man etwas Zeit und Energie sparen.
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 2 berechnet.
10lt Maische mit 8vol%:
A) Dreifachbrand:
Raubrand: 3lt mit 1.1 theor. Böden.
Mittelbrand: 1.5lt mit 1.1 theor. Böden.
Die Daten des Mittelbrands: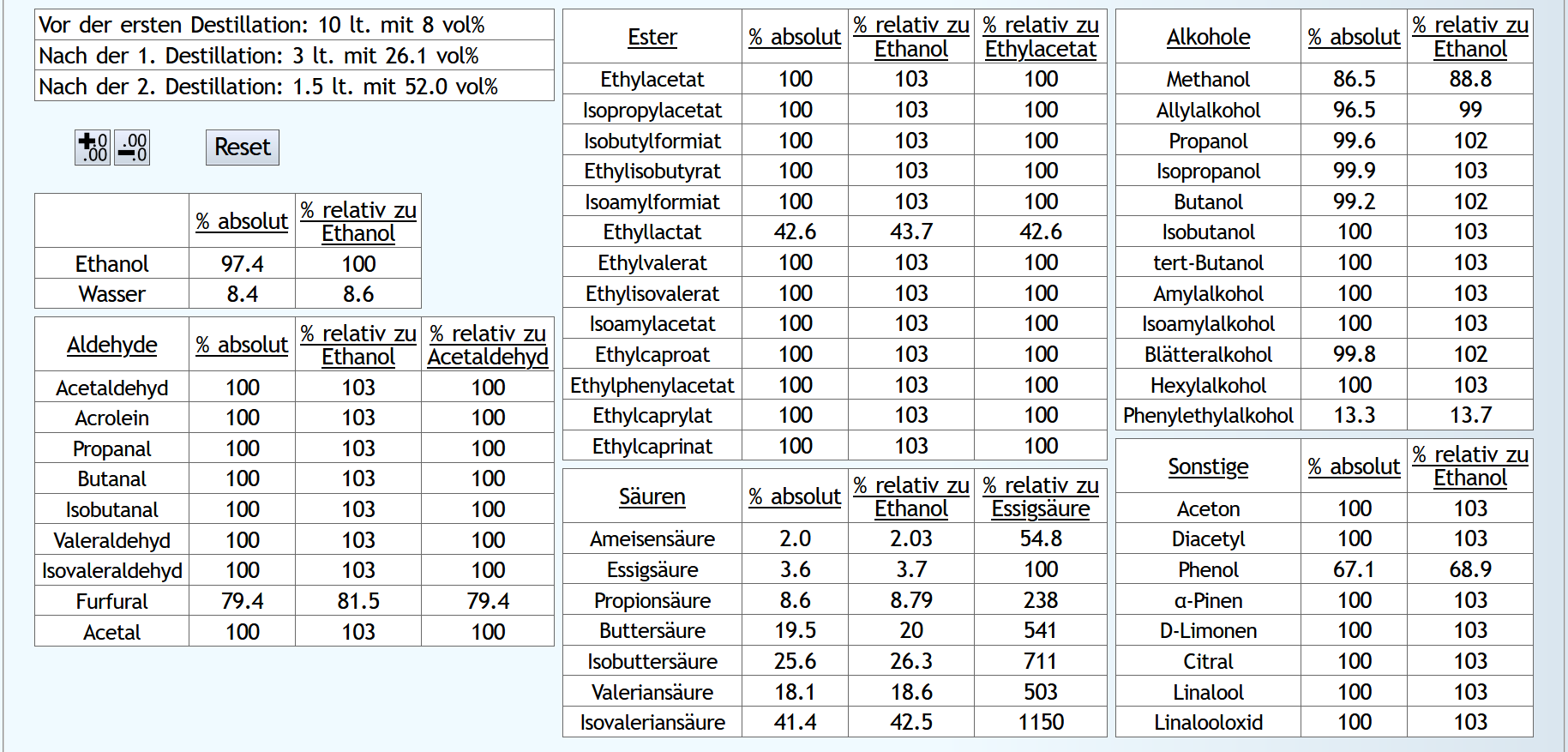 Ergibt 1.5lt mit 52vol%.
Ergibt 1.5lt mit 52vol%.
B) Doppelbrand:
Reflux-Raubrand bis ebenfalls 1.5lt 52vol%. Dafür sind 1.565 theor. Böden nötig.
Die Daten des Reflux-Raubrands: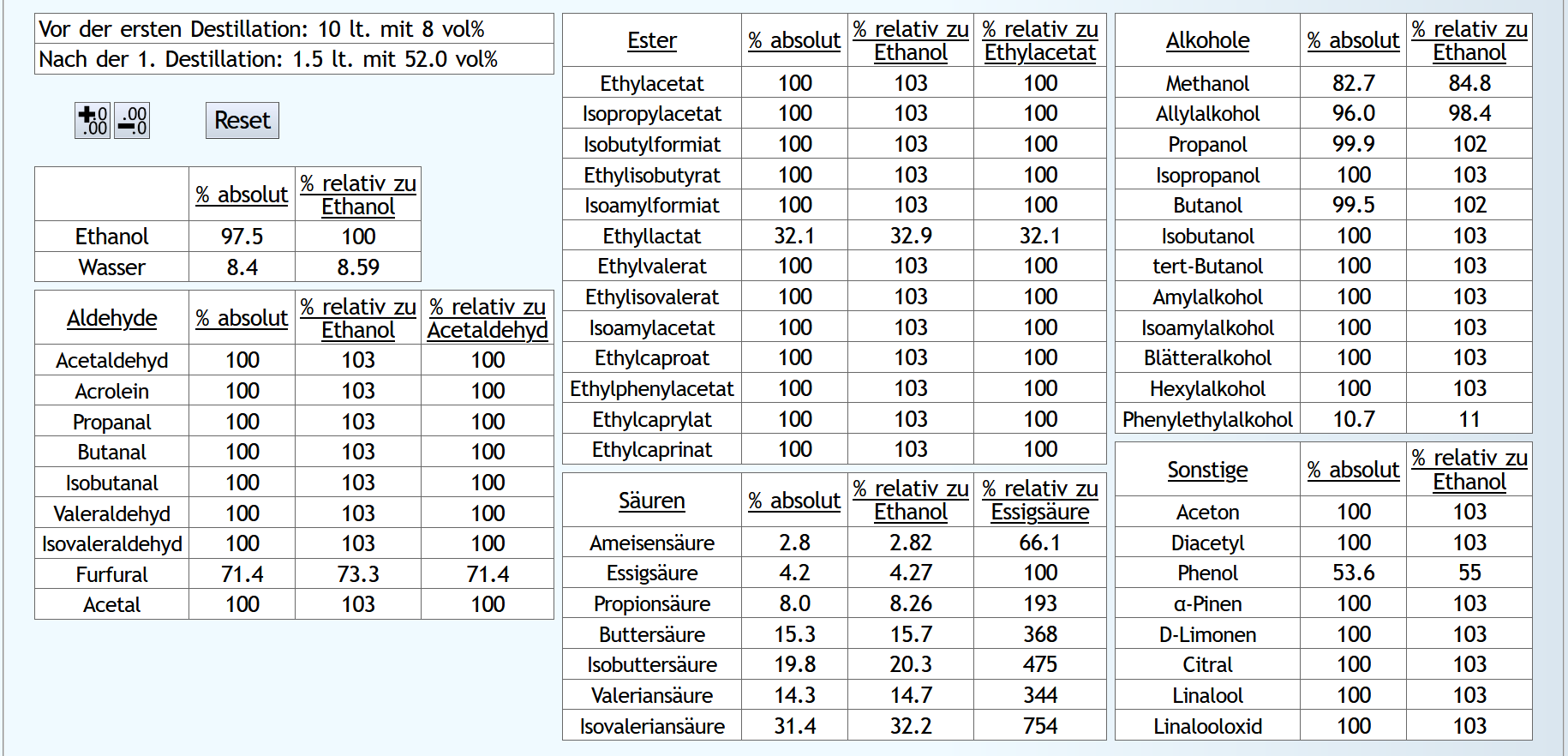 Bei beiden Versionen sind so gut wie alle Aldehyde, Ester, Alkohole und Sonstige im Destillat.
Beim Doppelbrand ein bisschen vollständiger, was tendenziell etwas besser ausschaut.
Aber bei den Säuren gibt es große Unterschiede.
Der Doppelbrand unterdrückt die Essigsäure und Ameisensäure besser, der Einfachbrand eher die größeren Säuren.
Da die Essigsäure bei weitem am häufigsten ist, wird der Doppelbrand etwas weicher sein.
Außerdem hat er mehr von den interessanten Säuren mit höherem Potential, welche beim Feinbrand dann vielleicht wertvolle Ester bilden.
Bei beiden Versionen sind so gut wie alle Aldehyde, Ester, Alkohole und Sonstige im Destillat.
Beim Doppelbrand ein bisschen vollständiger, was tendenziell etwas besser ausschaut.
Aber bei den Säuren gibt es große Unterschiede.
Der Doppelbrand unterdrückt die Essigsäure und Ameisensäure besser, der Einfachbrand eher die größeren Säuren.
Da die Essigsäure bei weitem am häufigsten ist, wird der Doppelbrand etwas weicher sein.
Außerdem hat er mehr von den interessanten Säuren mit höherem Potential, welche beim Feinbrand dann vielleicht wertvolle Ester bilden.
Nun könnte man den Refluxbrand aber auch mit ansteigendem Reflux machen, die Alkoholstärke also nie unter einen bestimmten Punkt fallen lassen.
C) Doppelbrand mit ansteigender Rektifikation:
Hier das Protokoll:
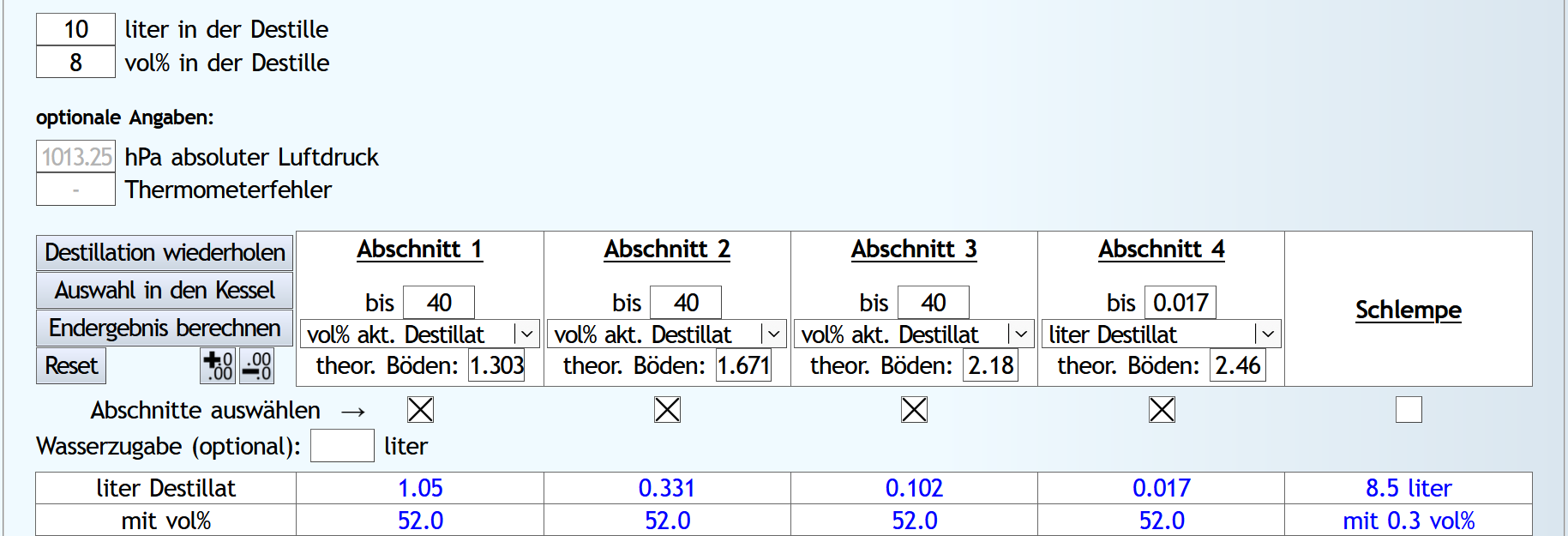 40vol% sollen also nicht unterschritten werden, am Ende sogar 45vol%.
Wegen der Vergleichbarkeit soll die gleiche Alkoholstärke und Destillatmenge wie bei A und B erreicht werden.
40vol% sollen also nicht unterschritten werden, am Ende sogar 45vol%.
Wegen der Vergleichbarkeit soll die gleiche Alkoholstärke und Destillatmenge wie bei A und B erreicht werden.
Das Endergebnis: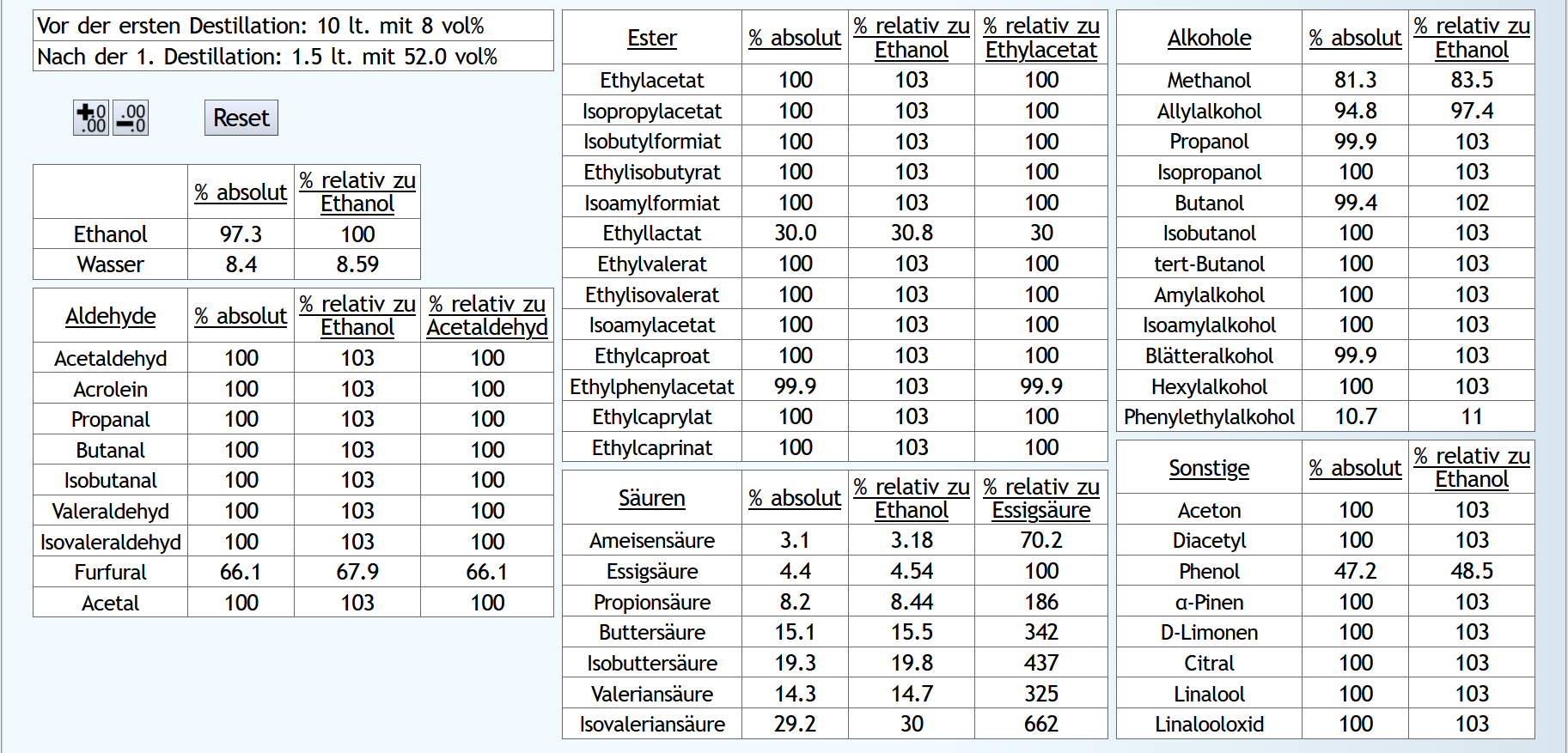 Wenn man B also schlechter als A findet (was wahrscheinlich ist), wird man C noch schlechter finden.
Alle an B kritisierten Punkte sind bei C noch etwas schlechter.
Wenn man B also schlechter als A findet (was wahrscheinlich ist), wird man C noch schlechter finden.
Alle an B kritisierten Punkte sind bei C noch etwas schlechter.
Fazit: Einen Rau- und Mittelbrand -beide unrektifiziert- durch einen Reflux-Raubrand zu ersetzen, bedeutet Qualitätseinbußen.
Die folgenden Beispiele sind mit dem Begleitstoffesimulator 2 berechnet.
10lt Maische mit 8vol%:
A) Dreifachbrand:
Raubrand: 3lt mit 1.1 theor. Böden.
Mittelbrand: 1.5lt mit 1.1 theor. Böden.
Die Daten des Mittelbrands:
B) Doppelbrand:
Reflux-Raubrand bis ebenfalls 1.5lt 52vol%. Dafür sind 1.565 theor. Böden nötig.
Die Daten des Reflux-Raubrands:
Nun könnte man den Refluxbrand aber auch mit ansteigendem Reflux machen, die Alkoholstärke also nie unter einen bestimmten Punkt fallen lassen.
C) Doppelbrand mit ansteigender Rektifikation:
Hier das Protokoll:
Das Endergebnis:
Fazit: Einen Rau- und Mittelbrand -beide unrektifiziert- durch einen Reflux-Raubrand zu ersetzen, bedeutet Qualitätseinbußen.
Hier die komplette Liste unserer Quellen
- "From pot stills to continuous stills: flavor modification by distillation" von Robert Piggot. Veröffentlicht in "The Alcohol Textbook", Kapitel 17
- "Whisky Technology, Production and Marketing", original von Panek und Boucher, wahrscheinlich "Continuous distillation", veröffentlicht wahrscheinlich auch in "The Science and Technology of Whiskies" von J.R.Piggott, Longman Scientific and Technical
- "Chemical aspects of distilling wines into brandy" von J.F. Guymon, 1973, veröffentlicht in "Chemistry of Winemaking"; Webb, A.; Advances in Chemistry; American Chemical Society: Washington, DC, 1974
- "Computer simulation applied to studying continuous spirit distillation and product quality control" von Fabio R.M. Batista, Antonio J.A. Meirelles, Food Control 22 (2011) 1592-1603
- "Vapor-liquid equilibria of organic homologues in ethanol-water solutions" von C.Williams
- "A quick method for obtaining partition factor of congeners in spirits" von A.Martin
- "Vapor-Liquid Equilibria Measurements of Bitter Orange Aroma Compounds Highly Diluted in Boiling Hydro-Alcoholic Solutions at 101.3 kPa" von S.Deterre
- "Distillation principles and processes" von S.Young, S.308
- "Vapor−Liquid Equilibrium of Ethyl Lactate Highly Diluted in Ethanol−Water Mixtures at 101.3 kPa. Experimental Measurements and Thermodynamic Modeling Using Semiempirical Models" von C.Puentes
- "Review and thermodynamic modeling with NRTL model of vapor–liquid equilibria (VLE) of aroma compounds highly diluted in ethanol–water mixtures at 101.3 kPa" von C.Puentes
- "Modélisation des équilibres entre phases et simulation de la distillation des eaux-de-vie en vue d’une meilleure compréhension du comportement des composés volatils d’arôme" von C.Puentes
- "Vapour–liquid equilibria of aroma compounds in hydroalcoholic solutions: Measurements with a recirculation method and modelling with the NRTL and COSMO-SAC approaches" von V.Athes
- "Vapor-Liquid Equilibria of a Minute Amount of n-Propyl, Isobutyl, and Isoamyl Alcohols in Aqueous Ethanol Solution under Reduced Pressure" von A.Ikari
- "Vapor-Liquid Equilibria of a Minute Amount of Methanol, Isovaleraldehyde and Diacetyl in Aqueous Ethanol Solution under Reduced Pressure" von A.Ikari
- "Behavior of a minute amount of furfural in distillation of aqueous ethanol solution under reduced pressure" von A.Ikari
- "Vapor-Liquid Equilibria of Trace Isobutyraldehyde, Ethyl Acetate and Isoamyl Acetate in Aqueous Ethanol Solution under Reduced Pressure" von A.Ikari
- "Review and thermodynamic modeling with NRTL model of vapor–liquid equilibria (VLE) of aroma compounds highly diluted in ethanol–water mixtures at 101.3 kPa" von C.Puentes
Die Reihenfolge ist zufällig.